

An integrated diagnosis strategy for congenital myopathies
- PMID: 23826317
- PMCID: PMC3691193
- DOI: 10.1371/journal.pone.0067527
An integrated diagnosis strategy for congenital myopathies
Abstract
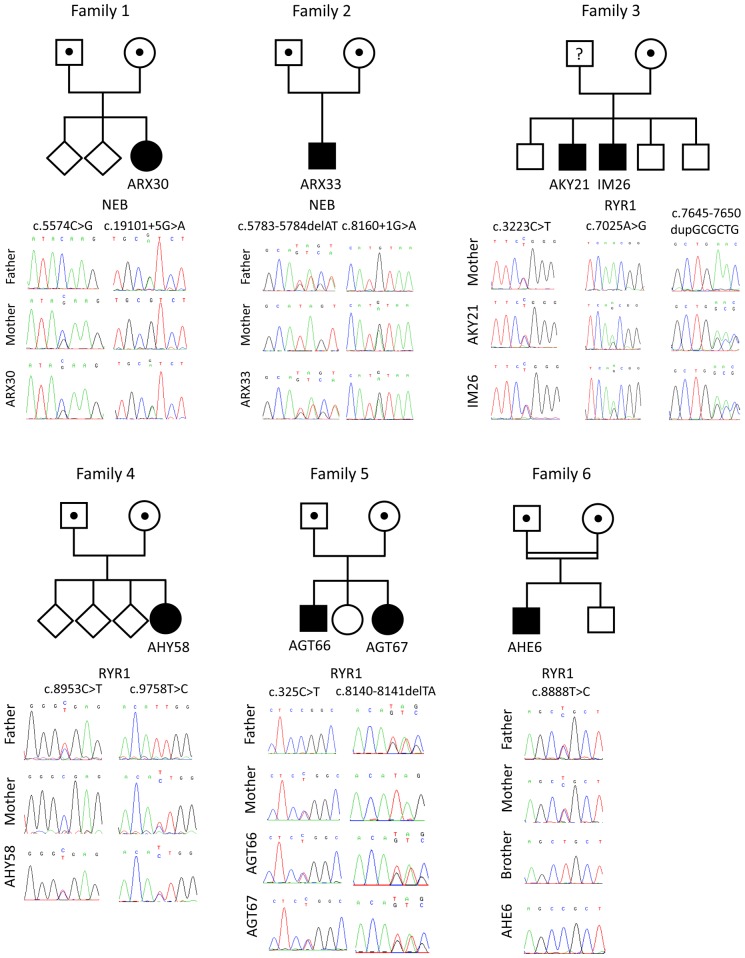
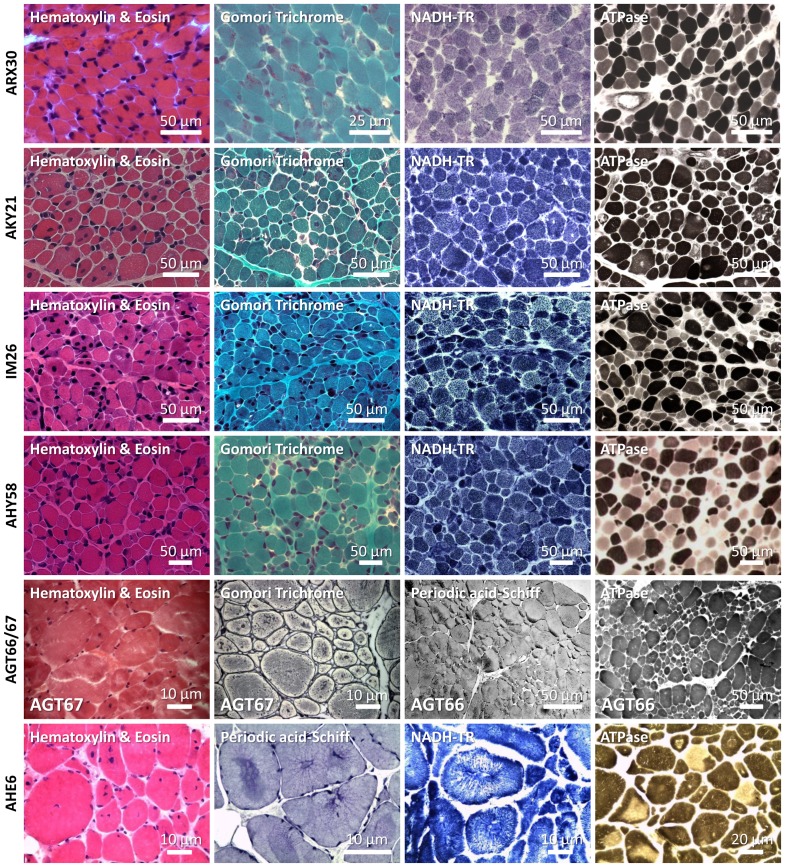
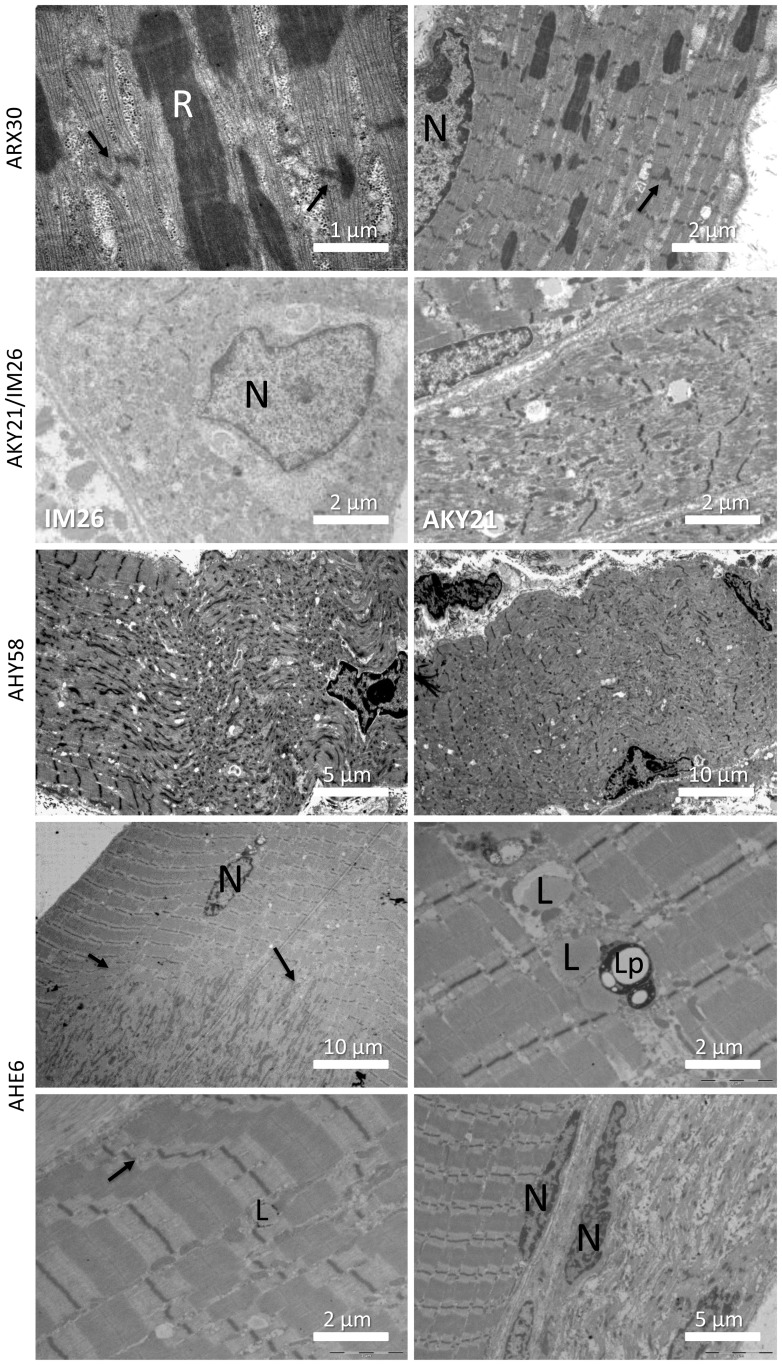
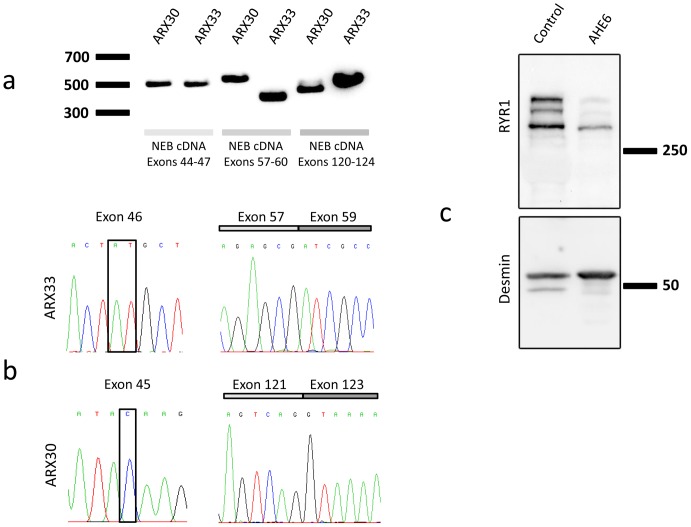
Congenital myopathies are severe muscle disorders affecting adults as well as children in all populations. The diagnosis of congenital myopathies is constrained by strong clinical and genetic heterogeneity. Moreover, the majority of patients present with unspecific histological features, precluding purposive molecular diagnosis and demonstrating the need for an alternative and more efficient diagnostic approach. We used exome sequencing complemented by histological and ultrastructural analysis of muscle biopsies to identify the causative mutations in eight patients with clinically different skeletal muscle pathologies, ranging from a fatal neonatal myopathy to a mild and slowly progressive myopathy with adult onset. We identified RYR1 (ryanodine receptor) mutations in six patients and NEB (nebulin) mutations in two patients. We found novel missense and nonsense mutations, unraveled small insertions/deletions and confirmed their impact on splicing and mRNA/protein stability. Histological and ultrastructural findings of the muscle biopsies of the patients validated the exome sequencing results. We provide the evidence that an integrated strategy combining exome sequencing with clinical and histopathological investigations overcomes the limitations of the individual approaches to allow a fast and efficient diagnosis, accelerating the patient's access to a better healthcare and disease management. This is of particular interest for the diagnosis of congenital myopathies, which involve very large genes like RYR1 and NEB as well as genetic and phenotypic heterogeneity.
Conflict of interest statement
Figures
References
-
- North K (2008) What's new in congenital myopathies? Neuromuscul Disord 18: 433–442. - PubMed
-
- Sewry CA (2008) Pathological defects in congenital myopathies. J Muscle Res Cell Motil 29: 231–238. - PubMed
-
- Amburgey K, McNamara N, Bennett LR, McCormick ME, Acsadi G, et al. (2011) Prevalence of congenital myopathies in a representative pediatric united states population. Ann Neurol 70: 662–665. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
