

A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection
- PMID: 23827683
- PMCID: PMC3763506
- DOI: 10.1016/j.cell.2013.06.013
A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection
Abstract
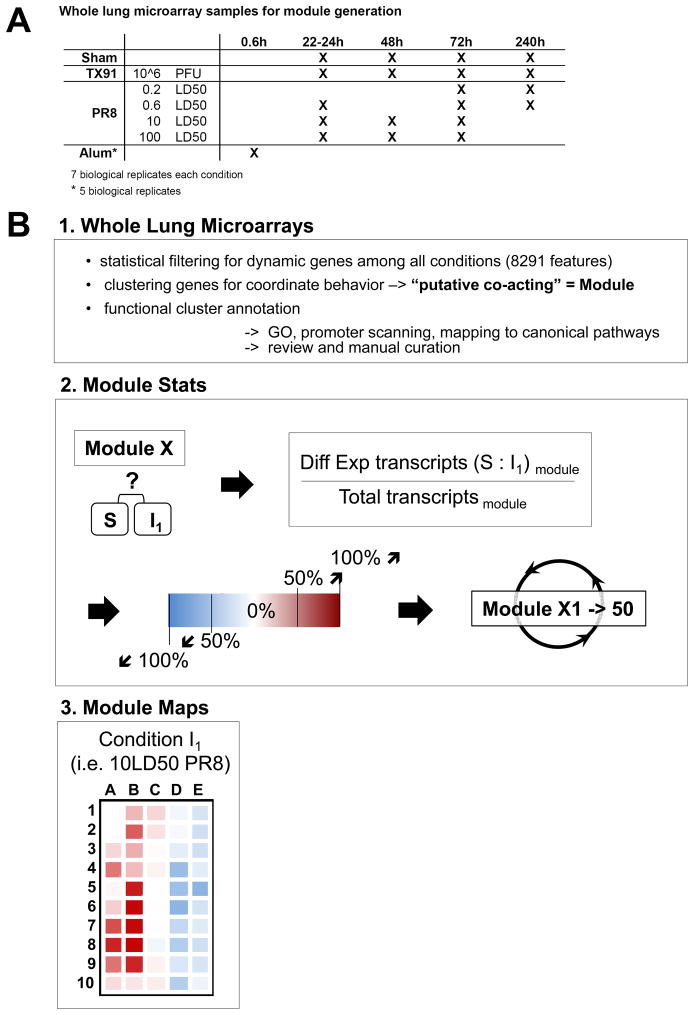
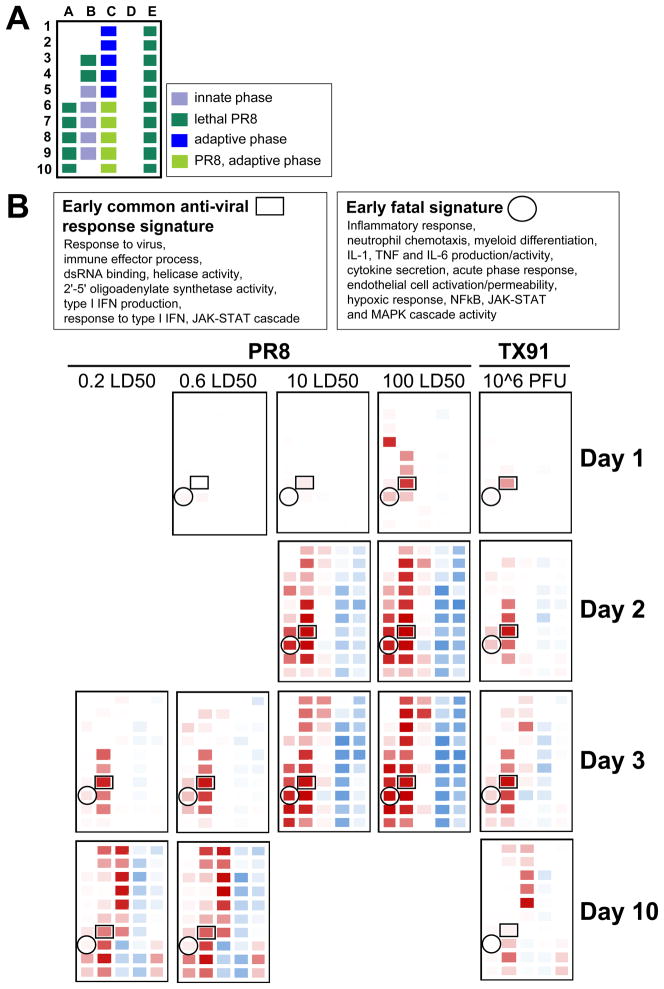
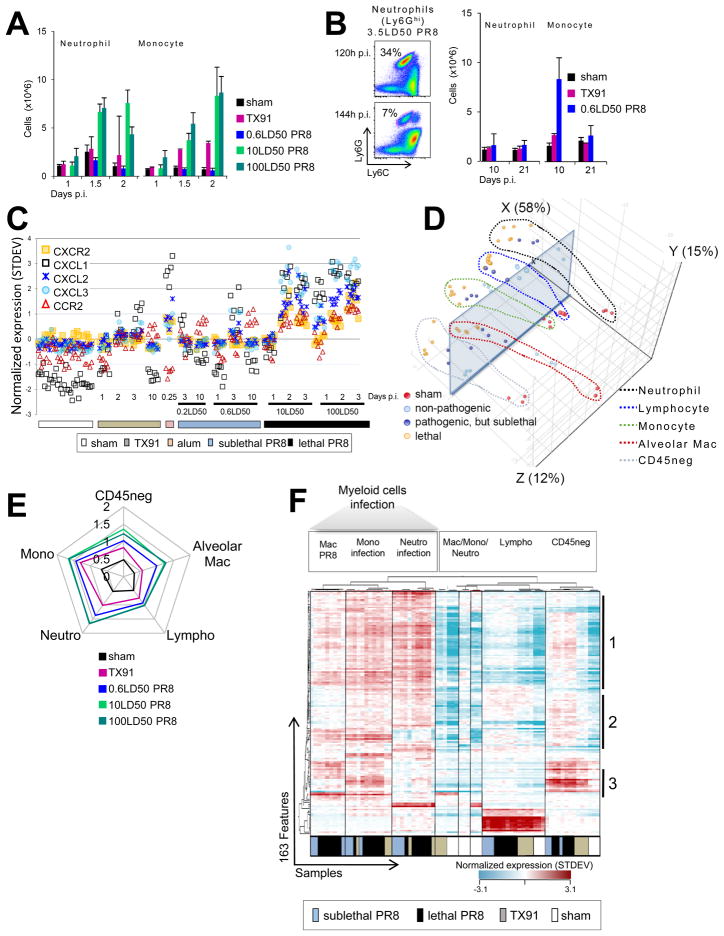
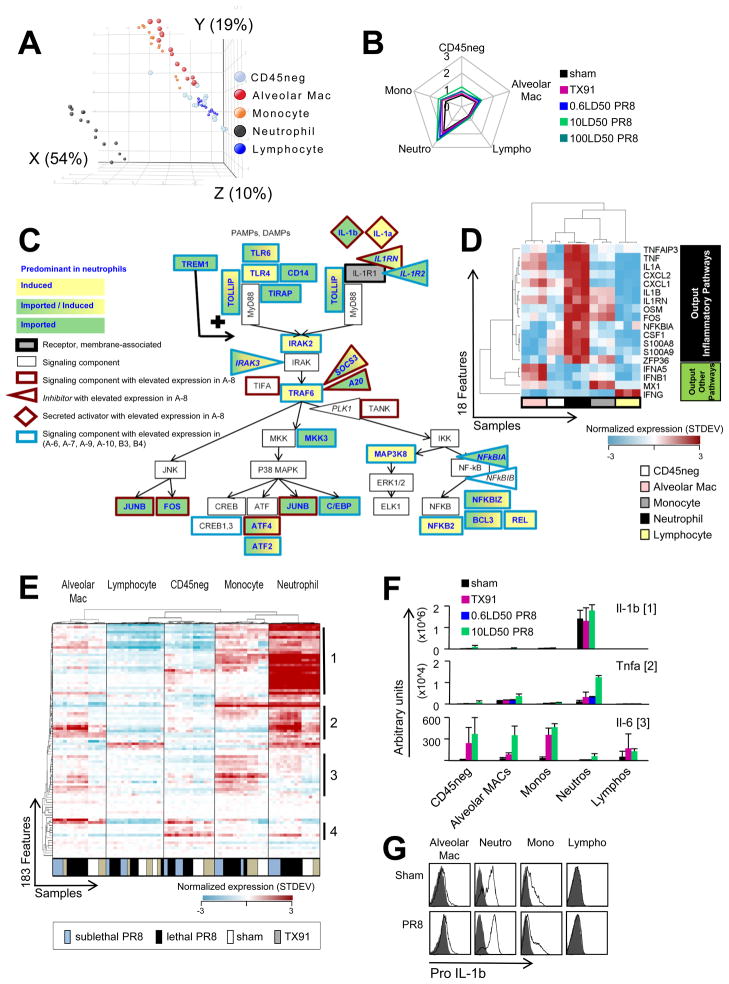
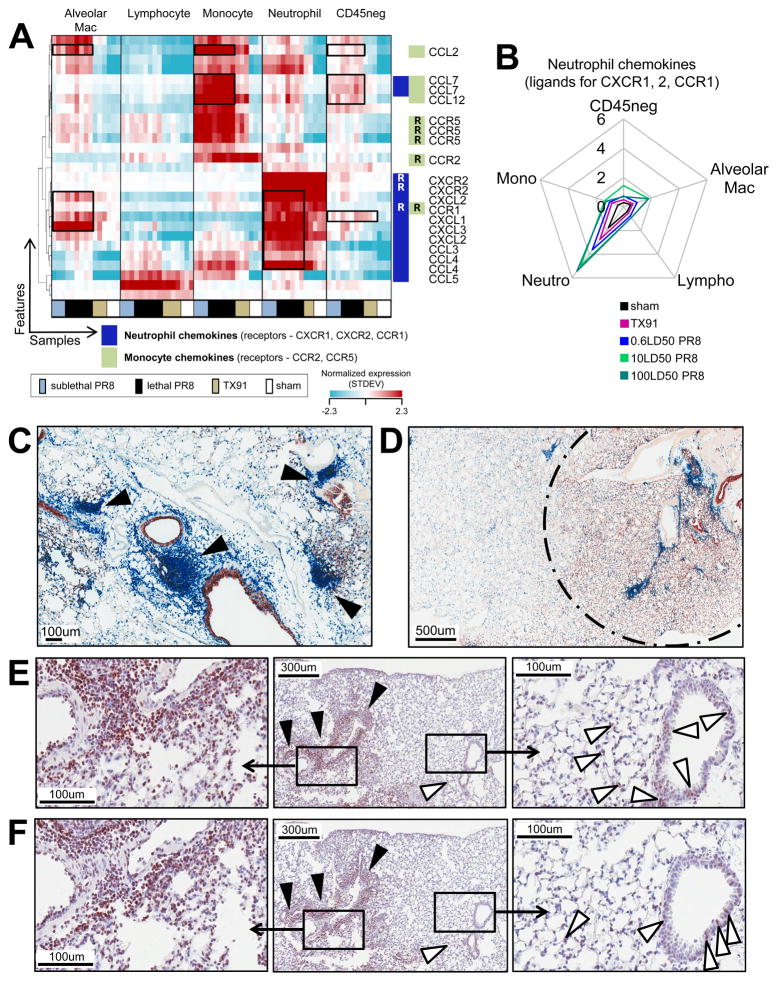
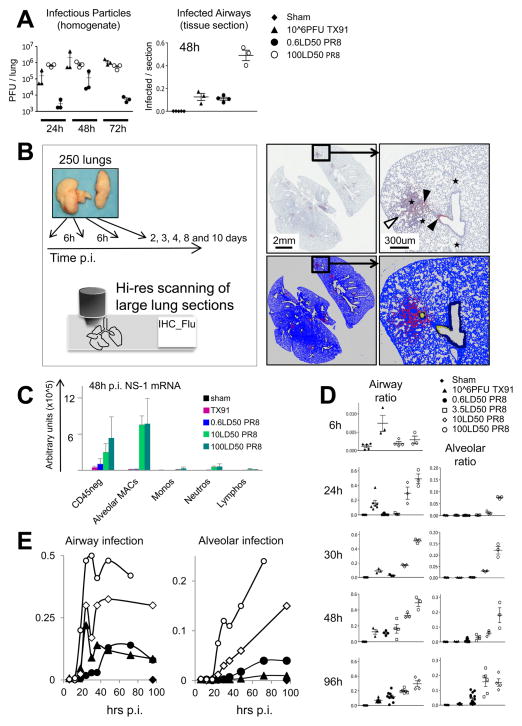
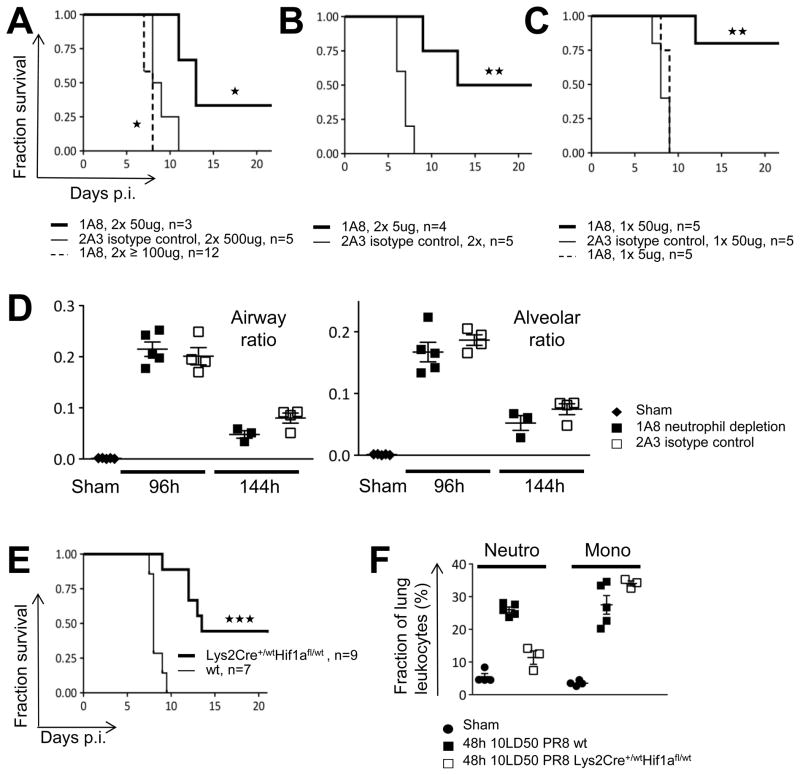
For acutely lethal influenza infections, the relative pathogenic contributions of direct viral damage to lung epithelium versus dysregulated immunity remain unresolved. Here, we take a top-down systems approach to this question. Multigene transcriptional signatures from infected lungs suggested that elevated activation of inflammatory signaling networks distinguished lethal from sublethal infections. Flow cytometry and gene expression analysis involving isolated cell subpopulations from infected lungs showed that neutrophil influx largely accounted for the predictive transcriptional signature. Automated imaging analysis, together with these gene expression and flow data, identified a chemokine-driven feedforward circuit involving proinflammatory neutrophils potently driven by poorly contained lethal viruses. Consistent with these data, attenuation, but not ablation, of the neutrophil-driven response increased survival without changing viral spread. These findings establish the primacy of damaging innate inflammation in at least some forms of influenza-induced lethality and provide a roadmap for the systematic dissection of infection-associated pathology.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Influenza virus: lethal weapons.Nat Rev Immunol. 2013 Aug;13(8):543. doi: 10.1038/nri3502. Epub 2013 Jul 19. Nat Rev Immunol. 2013. PMID: 23868220 No abstract available.
References
-
- Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, Shaw M, Uyeki TM, Zaki SR, Hayden FG, Hui DS, Kettner JD, et al. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med. 2010;362:1708–1719. - PubMed
-
- Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, Lochindarat S, Nguyen TK, Nguyen TH, Tran TH, et al. Avian influenza A (H5N1) infection in humans. N Engl J Med. 2005;353:1374–1385. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
