

MiST: a new approach to variant detection in deep sequencing datasets
- PMID: 23828039
- PMCID: PMC3763541
- DOI: 10.1093/nar/gkt551
MiST: a new approach to variant detection in deep sequencing datasets
Abstract
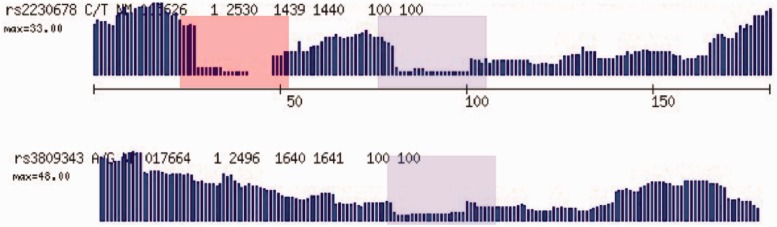
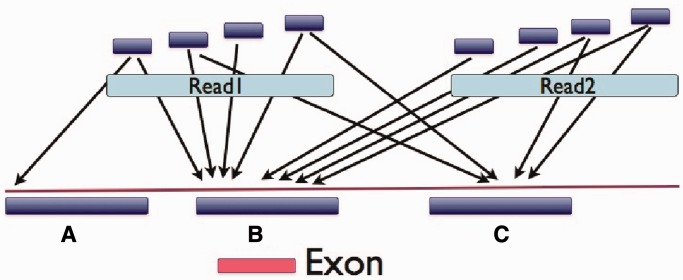
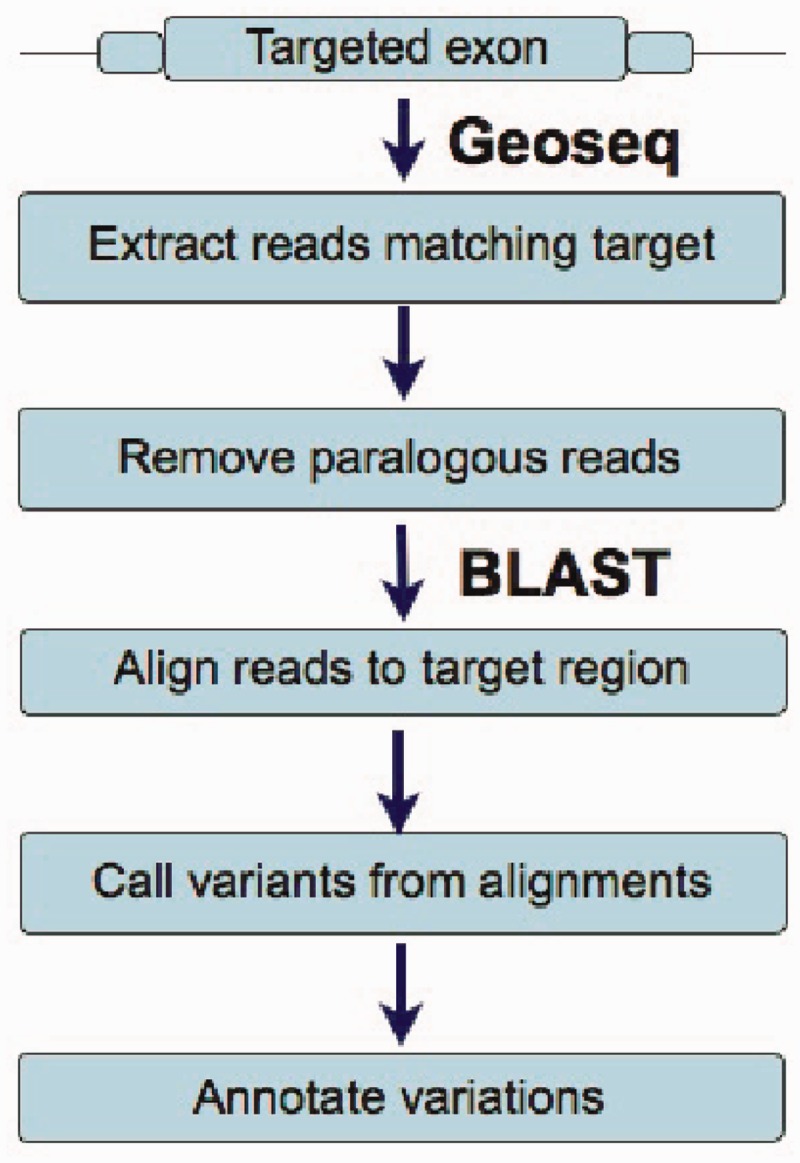
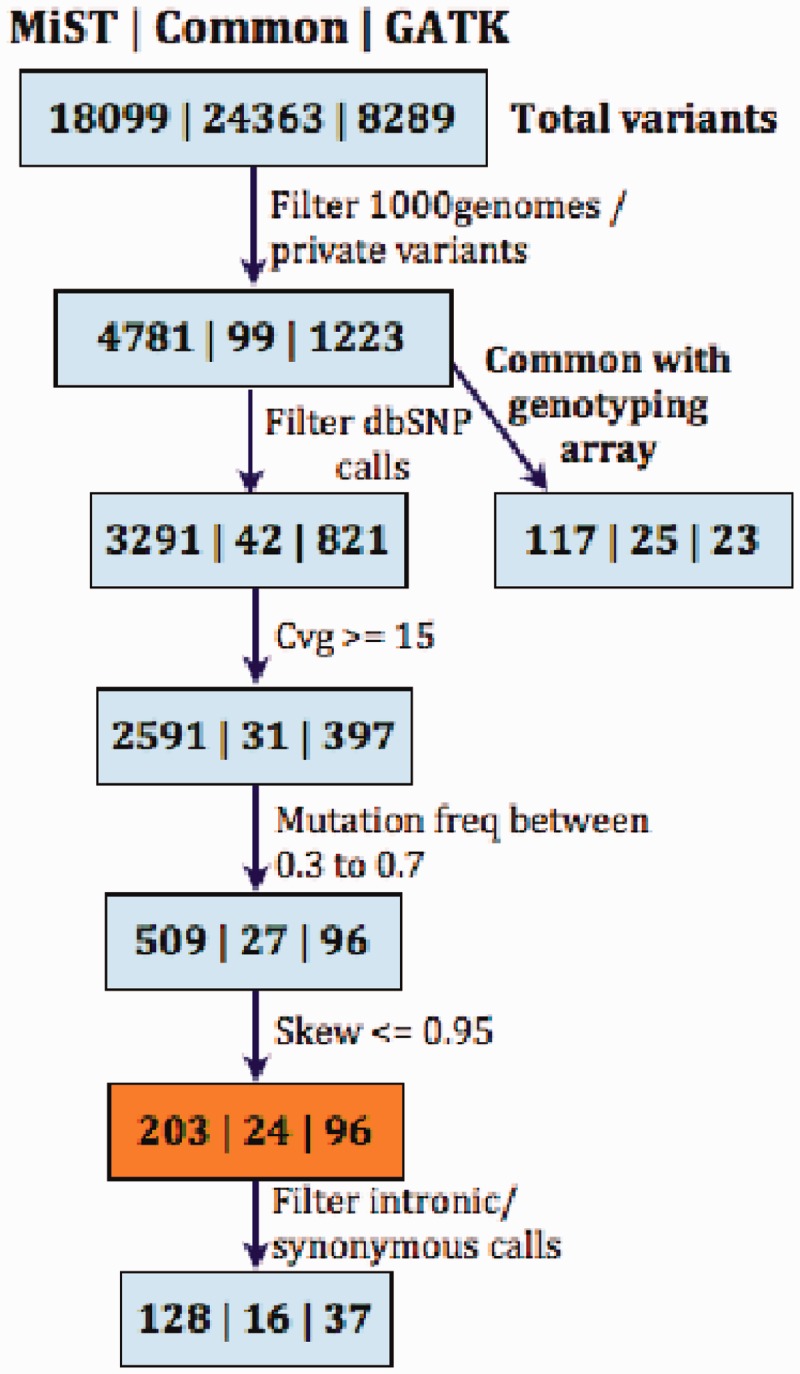
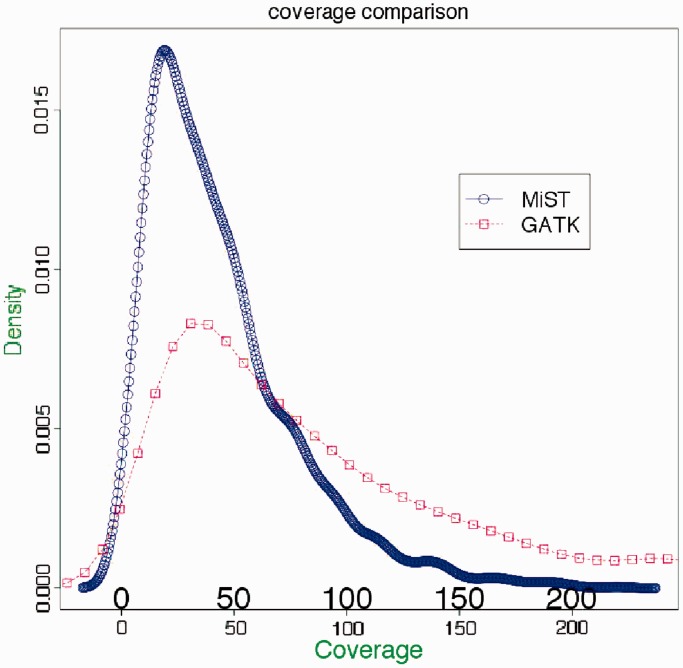
MiST is a novel approach to variant calling from deep sequencing data, using the inverted mapping approach developed for Geoseq. Reads that can map to a targeted exonic region are identified using exact matches to tiles from the region. The reads are then aligned to the targets to discover variants. MiST carefully handles paralogous reads that map ambiguously to the genome and clonal reads arising from PCR bias, which are the two major sources of errors in variant calling. The reduced computational complexity of mapping selected reads to targeted regions of the genome improves speed, specificity and sensitivity of variant detection. Compared with variant calls from the GATK platform, MiST showed better concordance with SNPs from dbSNP and genotypes determined by an exonic-SNP array. Variant calls made only by MiST confirm at a high rate (>90%) by Sanger sequencing. Thus, MiST is a valuable alternative tool to analyse variants in deep sequencing data.
Figures


References
-
- FreeBayes - the MarthLab. http://bioinformatics.bc.edu/marthlab/FreeBayes.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
