

R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli
- PMID: 23828459
- PMCID: PMC3715873
- DOI: 10.1038/ncomms3115
R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli
Erratum in
- Nat Commun. 2014;5:2762
Abstract
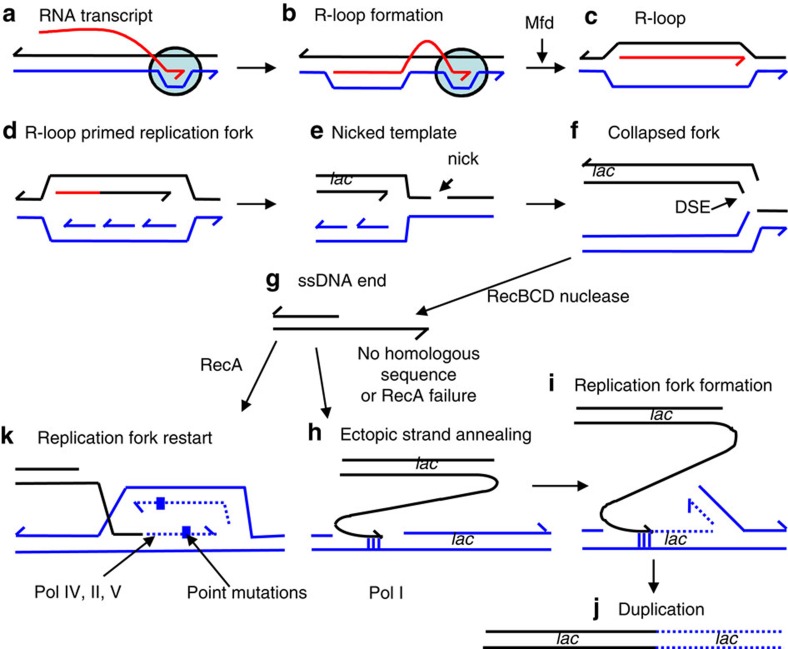
Double-stranded DNA ends, often from replication, drive genomic instability, yet their origin in non-replicating cells is unknown. Here we show that transcriptional RNA/DNA hybrids (R-loops) generate DNA ends that underlie stress-induced mutation and amplification. Depleting RNA/DNA hybrids with overproduced RNase HI reduces both genomic changes, indicating RNA/DNA hybrids as intermediates in both. An Mfd requirement and inhibition by translation implicate transcriptional R-loops. R-loops promote instability by generating DNA ends, shown by their dispensability when ends are provided by I-SceI endonuclease. Both R-loops and single-stranded endonuclease TraI are required for end formation, visualized as foci of a fluorescent end-binding protein. The data suggest that R-loops prime replication forks that collapse at single-stranded nicks, producing ends that instigate genomic instability. The results illuminate how DNA ends form in non-replicating cells, identify R-loops as the earliest known mutation/amplification intermediate, and suggest that genomic instability during stress could be targeted to transcribed regions, accelerating adaptation.
Figures
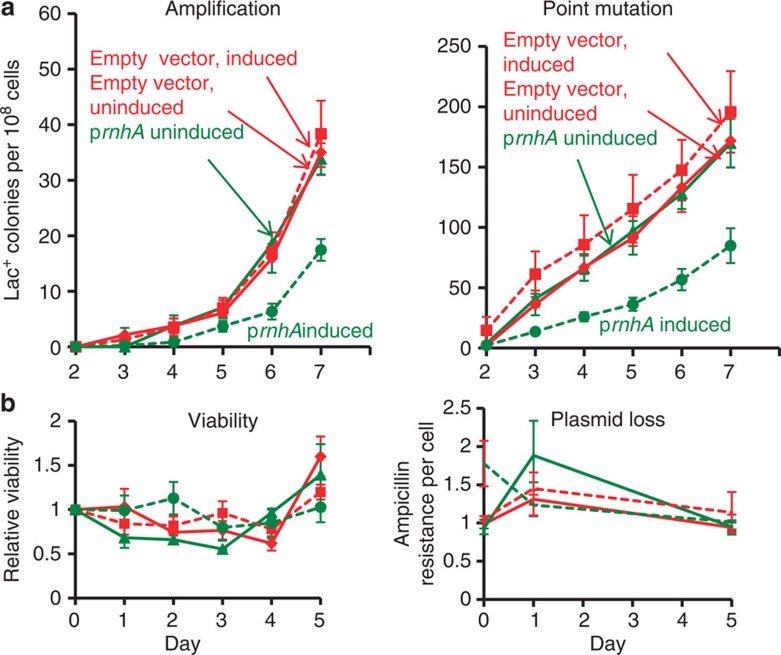
 ) (solid line) or induced (
) (solid line) or induced ( ) (broken line) by the presence of arabinose. Also strain PJH1091, carries empty vector pBAD18 as a control, with transcription from the PBAD promoter induced (♦) or uninduced (
) (broken line) by the presence of arabinose. Also strain PJH1091, carries empty vector pBAD18 as a control, with transcription from the PBAD promoter induced (♦) or uninduced ( ) by presence or absence of arabinose respectively. (b) Viability and retention of pBAD18-rnhA by day 5 of the experiment shows no plasmid loss. Viability and plasmid loss plots are offset by 2 days: the time needed to form a visible Lac+ colony. Error bars, one s.e.m. of four parallel cultures. These and all experiments were performed three times with comparable results.
) by presence or absence of arabinose respectively. (b) Viability and retention of pBAD18-rnhA by day 5 of the experiment shows no plasmid loss. Viability and plasmid loss plots are offset by 2 days: the time needed to form a visible Lac+ colony. Error bars, one s.e.m. of four parallel cultures. These and all experiments were performed three times with comparable results.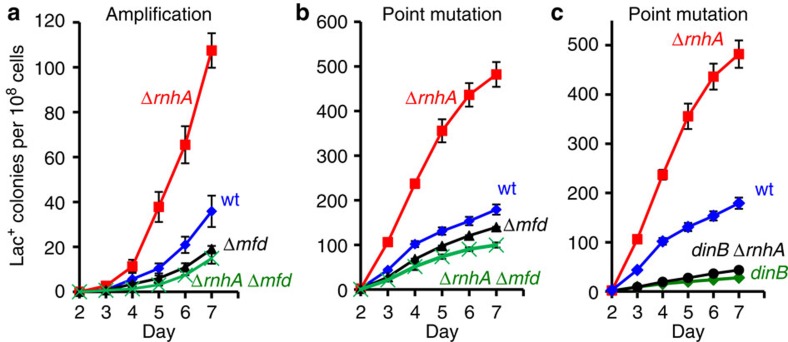 ), mfd (), and mfd ΔrnhA ( × ) strains SMR4562, PJH683, PJH813 and PJH946. (c) Increased point mutation in cells lacking RNase HI requires DinB/DNA Pol IV, as in RNase HI-proficient cells. Wild-type (♦), ΔrnhA (), dinB10 (♦), dinB10 ΔrnhA () strains SMR4562, PJH683, SMR5830 and PJH791. Error bars, one s.e.m. of four parallel cultures. These experiments were performed three times with comparable results.
), mfd (), and mfd ΔrnhA ( × ) strains SMR4562, PJH683, PJH813 and PJH946. (c) Increased point mutation in cells lacking RNase HI requires DinB/DNA Pol IV, as in RNase HI-proficient cells. Wild-type (♦), ΔrnhA (), dinB10 (♦), dinB10 ΔrnhA () strains SMR4562, PJH683, SMR5830 and PJH791. Error bars, one s.e.m. of four parallel cultures. These experiments were performed three times with comparable results.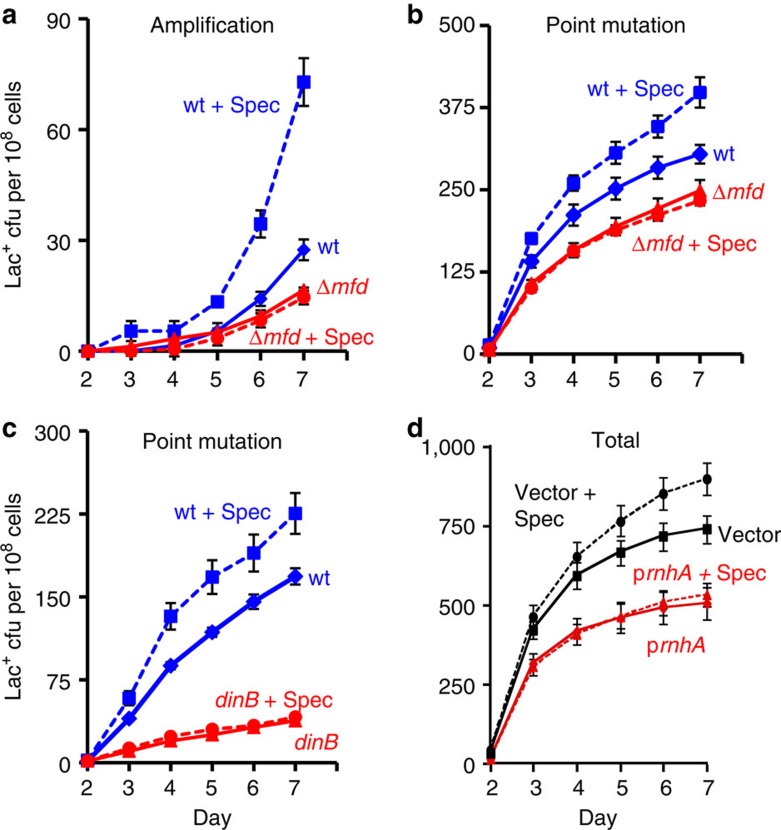 ), WT (♦), mfd+spec (), mfd (). (c) The spectinomycin treatment-induced increase in point mutagenesis is DinB/Pol IV-dependent. Point mutation in WT (SMR4562) and dinB (SMR5830) cells pulsed with spec. WT+spec (), WT (♦), dinB+spec (), dinB (). The curves for the spectinomycin-treated WT cells differ from those of the untreated WT cells significantly, WT and spec-treated WT differ (for three experiments, P=0.002 for point mutation and 0.001 for amplification, Student’s t-test); mfd and spec-treated mfd are not significantly different (P=0.6 for point mutation, and 0.7 for amplification). (d) Spectinomycin treatment does not increase Lac+ revertants in cells overproducing RNase HI. Vector pBAD18 (), vector pBAD18 spec-treated (), pBAD18-rnhA (), pBAD18-rnhA spec-treated (); broken lines denote spectinomycin treatment. Error bars represent one s.e.m. of four parallel cultures. These experiments were performed three times with comparable results.
), WT (♦), mfd+spec (), mfd (). (c) The spectinomycin treatment-induced increase in point mutagenesis is DinB/Pol IV-dependent. Point mutation in WT (SMR4562) and dinB (SMR5830) cells pulsed with spec. WT+spec (), WT (♦), dinB+spec (), dinB (). The curves for the spectinomycin-treated WT cells differ from those of the untreated WT cells significantly, WT and spec-treated WT differ (for three experiments, P=0.002 for point mutation and 0.001 for amplification, Student’s t-test); mfd and spec-treated mfd are not significantly different (P=0.6 for point mutation, and 0.7 for amplification). (d) Spectinomycin treatment does not increase Lac+ revertants in cells overproducing RNase HI. Vector pBAD18 (), vector pBAD18 spec-treated (), pBAD18-rnhA (), pBAD18-rnhA spec-treated (); broken lines denote spectinomycin treatment. Error bars represent one s.e.m. of four parallel cultures. These experiments were performed three times with comparable results.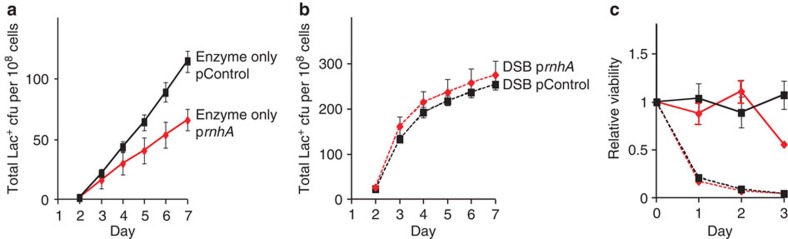 ). (b) The DSB-producing strain (carrying the chromosomal inducible I-SceI enzyme and I-SceI cutsite near lac) that over-produces RNase HI (PJH1321) (♦) shows no reduction of Lac+ mutants compared with the isogenic strain carrying the control plasmid (PJH1319) (), indicating that R-loops are not necessary for mutation when DSBs are provided. This result holds when both I-SceI enzyme and cutsite are present and not in strains with the enzyme only (a). (c) Strains experiencing double-strand cutting by I-SceI show reduced viability. However, the viability decrease is the same for cells with the rnhA plasmid as for those with the control plasmid making quantitative comparisons meaningful. Relative viability was determined per. Values in (a,b) have not been corrected for declining viability. Error bars represent one s.e.m. of four parallel cultures. Each experiment was performed three times with comparable results.
). (b) The DSB-producing strain (carrying the chromosomal inducible I-SceI enzyme and I-SceI cutsite near lac) that over-produces RNase HI (PJH1321) (♦) shows no reduction of Lac+ mutants compared with the isogenic strain carrying the control plasmid (PJH1319) (), indicating that R-loops are not necessary for mutation when DSBs are provided. This result holds when both I-SceI enzyme and cutsite are present and not in strains with the enzyme only (a). (c) Strains experiencing double-strand cutting by I-SceI show reduced viability. However, the viability decrease is the same for cells with the rnhA plasmid as for those with the control plasmid making quantitative comparisons meaningful. Relative viability was determined per. Values in (a,b) have not been corrected for declining viability. Error bars represent one s.e.m. of four parallel cultures. Each experiment was performed three times with comparable results.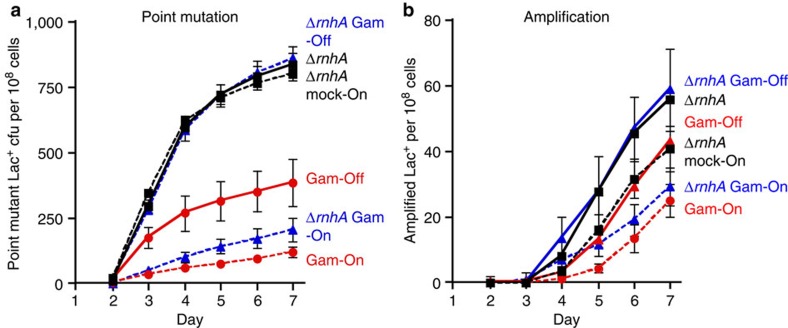 ); ΔrnhA PN25tetOgam tetR, PJH2443 (); PN25tetOgam tetR, PJH2023 ().Gam-On induction, broken lines. Gam-Off, the same strains as ‘Gam-On’ without doxycycline induction; mock-On, strains with the Tet repressor but without the gam gene treated with doxycycline. Error bars represent one s.e.m. of four parallel cultures. This experiment was performed three times with comparable results.
); ΔrnhA PN25tetOgam tetR, PJH2443 (); PN25tetOgam tetR, PJH2023 ().Gam-On induction, broken lines. Gam-Off, the same strains as ‘Gam-On’ without doxycycline induction; mock-On, strains with the Tet repressor but without the gam gene treated with doxycycline. Error bars represent one s.e.m. of four parallel cultures. This experiment was performed three times with comparable results.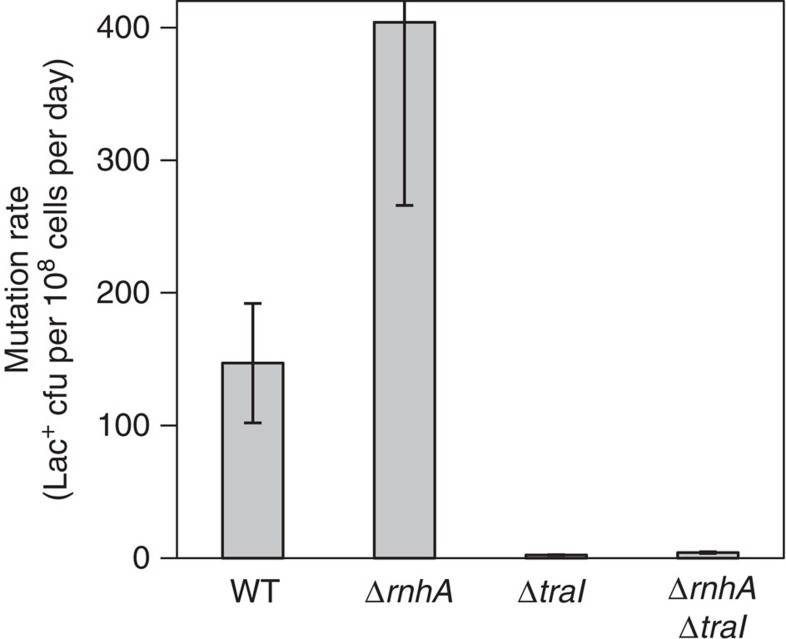
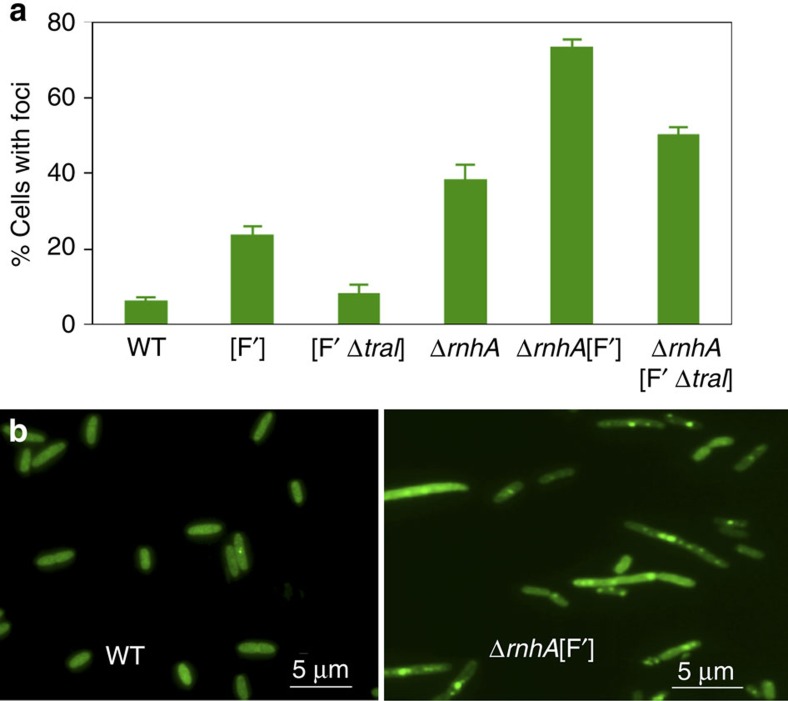
Similar articles
-
The RecG branch migration protein of Escherichia coli dissociates R-loops.J Mol Biol. 1996 Dec 13;264(4):713-21. doi: 10.1006/jmbi.1996.0671. J Mol Biol. 1996. PMID: 8980680
-
Genome-wide Map of R-Loop-Induced Damage Reveals How a Subset of R-Loops Contributes to Genomic Instability.Mol Cell. 2018 Aug 16;71(4):487-497.e3. doi: 10.1016/j.molcel.2018.06.037. Epub 2018 Aug 2. Mol Cell. 2018. PMID: 30078723 Free PMC article.
-
Toxic R-loops: Cause or consequence of replication stress?DNA Repair (Amst). 2021 Nov;107:103199. doi: 10.1016/j.dnarep.2021.103199. Epub 2021 Aug 3. DNA Repair (Amst). 2021. PMID: 34399314 Review.
-
Interaction with single-stranded DNA-binding protein localizes ribonuclease HI to DNA replication forks and facilitates R-loop removal.Mol Microbiol. 2020 Sep;114(3):495-509. doi: 10.1111/mmi.14529. Epub 2020 Jun 4. Mol Microbiol. 2020. PMID: 32426857 Free PMC article.
-
The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability.DNA Repair (Amst). 2014 Jul;19:84-94. doi: 10.1016/j.dnarep.2014.03.023. Epub 2014 Apr 18. DNA Repair (Amst). 2014. PMID: 24746923 Free PMC article. Review.
Cited by
-
DNA damage, inflammation and aging: Insights from mice.Front Aging. 2022 Sep 7;3:973781. doi: 10.3389/fragi.2022.973781. eCollection 2022. Front Aging. 2022. PMID: 36160606 Free PMC article. Review.
-
An underlying mechanism for the increased mutagenesis of lagging-strand genes in Bacillus subtilis.Proc Natl Acad Sci U S A. 2015 Mar 10;112(10):E1096-105. doi: 10.1073/pnas.1416651112. Epub 2015 Feb 23. Proc Natl Acad Sci U S A. 2015. PMID: 25713353 Free PMC article.
-
R-loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias.PLoS One. 2014 Mar 17;9(3):e90219. doi: 10.1371/journal.pone.0090219. eCollection 2014. PLoS One. 2014. PMID: 24637776 Free PMC article.
-
Engineered proteins detect spontaneous DNA breakage in human and bacterial cells.Elife. 2013 Oct 29;2:e01222. doi: 10.7554/eLife.01222. Elife. 2013. PMID: 24171103 Free PMC article.
-
DNA Breaks-Mediated Fitness Cost Reveals RNase HI as a New Target for Selectively Eliminating Antibiotic-Resistant Bacteria.Mol Biol Evol. 2021 Jul 29;38(8):3220-3234. doi: 10.1093/molbev/msab093. Mol Biol Evol. 2021. PMID: 33830249 Free PMC article.
References
-
- Cousineau I., Abaji C. & Belmaaza A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: implications for sister chromatid cohesion, genome stability and carcinogenesis. Cancer Res. 65, 11384–11391 (2005). - PubMed
-
- Debacker K. & Kooy R. F. Fragile sites and human disease. Hum. Mol. Genet. 16, Spec no. 2R150–R158 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
