

Scalable metagenomic taxonomy classification using a reference genome database
- PMID: 23828782
- PMCID: PMC3753567
- DOI: 10.1093/bioinformatics/btt389
Scalable metagenomic taxonomy classification using a reference genome database
Abstract
Motivation: Deep metagenomic sequencing of biological samples has the potential to recover otherwise difficult-to-detect microorganisms and accurately characterize biological samples with limited prior knowledge of sample contents. Existing metagenomic taxonomic classification algorithms, however, do not scale well to analyze large metagenomic datasets, and balancing classification accuracy with computational efficiency presents a fundamental challenge.
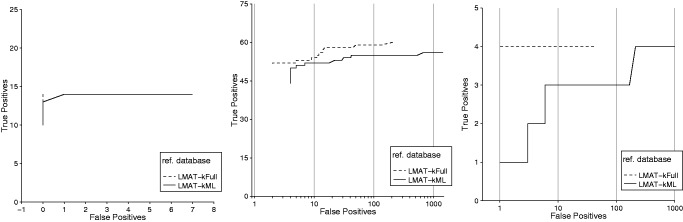
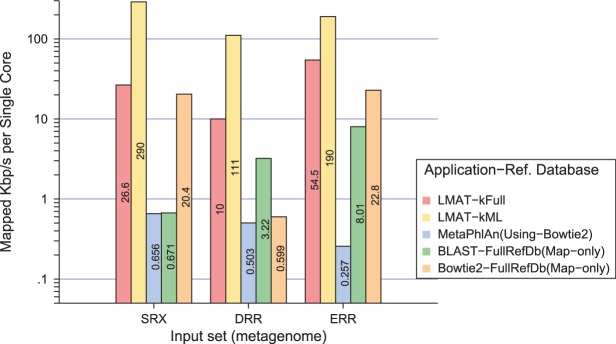
Results: A method is presented to shift computational costs to an off-line computation by creating a taxonomy/genome index that supports scalable metagenomic classification. Scalable performance is demonstrated on real and simulated data to show accurate classification in the presence of novel organisms on samples that include viruses, prokaryotes, fungi and protists. Taxonomic classification of the previously published 150 giga-base Tyrolean Iceman dataset was found to take <20 h on a single node 40 core large memory machine and provide new insights on the metagenomic contents of the sample.
Availability: Software was implemented in C++ and is freely available at http://sourceforge.net/projects/lmat
Contact: allen99@llnl.gov
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
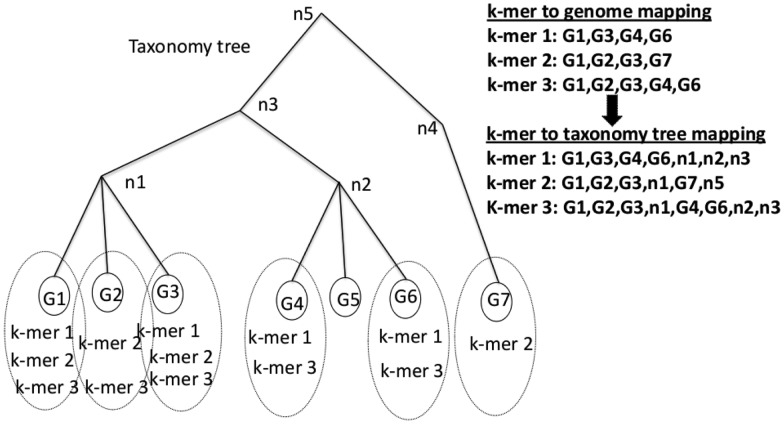
 , and leaf node genomes
, and leaf node genomes  , all of which are labeled with taxonomy IDs. k-mers (k-mer1, k-mer2, k-mer3) are linked to their source genomes (dotted circles) and their taxonomy hierarchy up to the LCA
, all of which are labeled with taxonomy IDs. k-mers (k-mer1, k-mer2, k-mer3) are linked to their source genomes (dotted circles) and their taxonomy hierarchy up to the LCA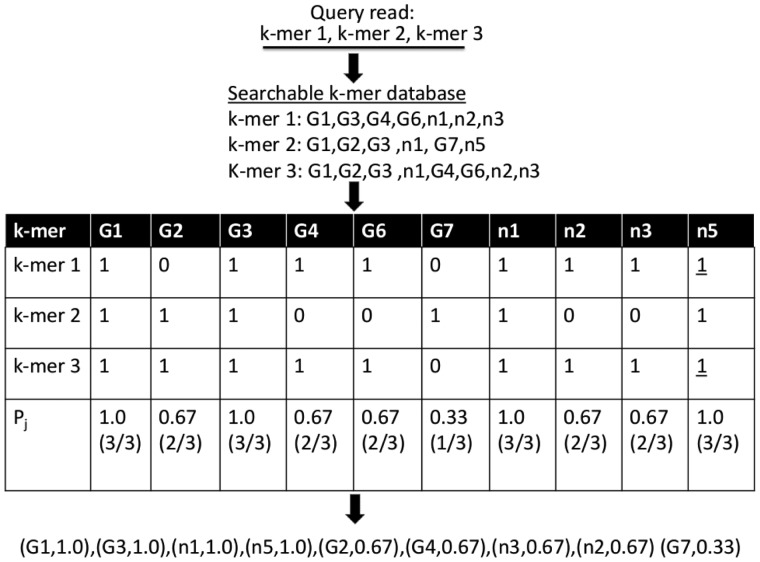
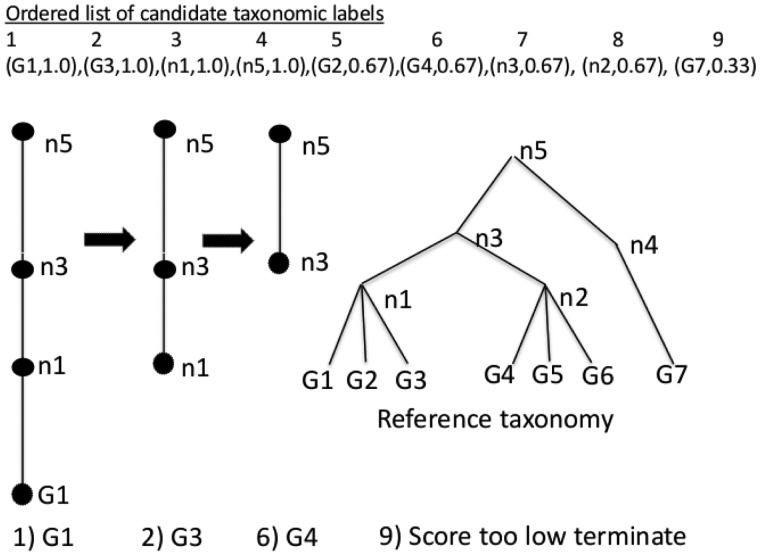
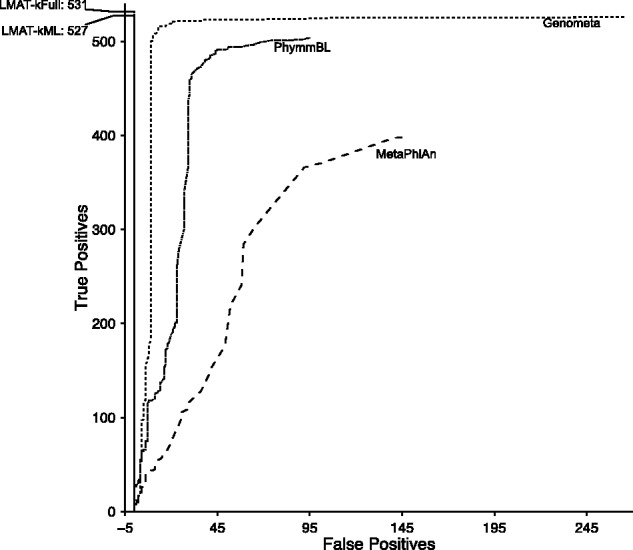
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
