

Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability
- PMID: 23830518
- PMCID: PMC3710757
- DOI: 10.1016/j.ajhg.2013.05.028
Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability
Abstract
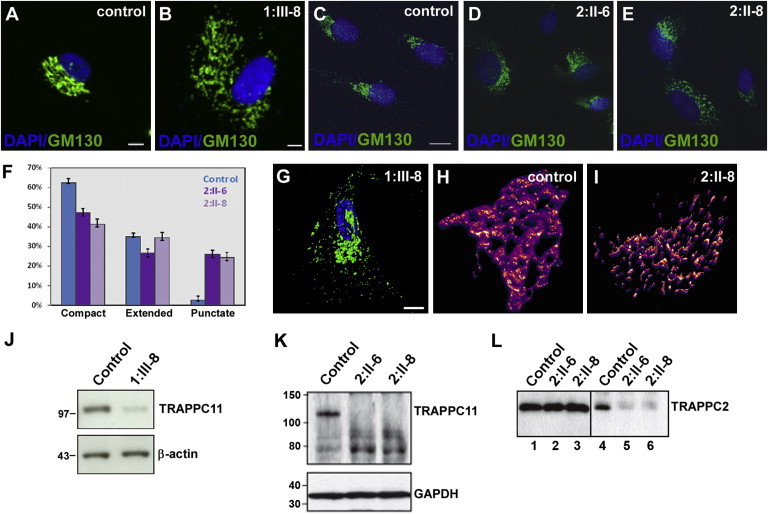
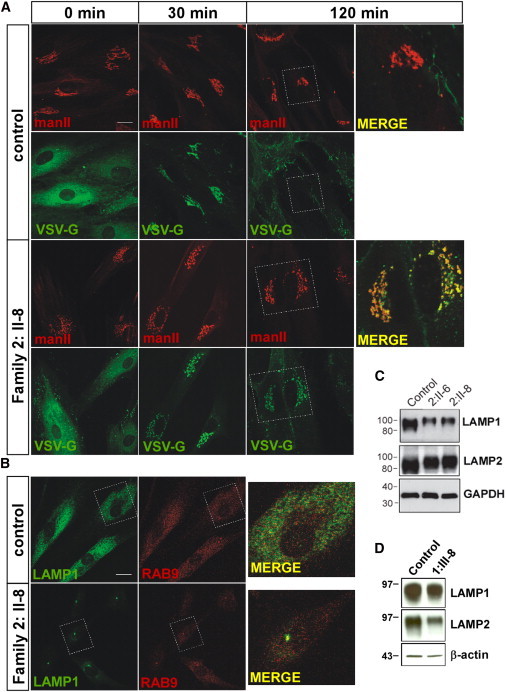
Myopathies are a clinically and etiologically heterogeneous group of disorders that can range from limb girdle muscular dystrophy (LGMD) to syndromic forms with associated features including intellectual disability. Here, we report the identification of mutations in transport protein particle complex 11 (TRAPPC11) in three individuals of a consanguineous Syrian family presenting with LGMD and in five individuals of Hutterite descent presenting with myopathy, infantile hyperkinetic movements, ataxia, and intellectual disability. By using a combination of whole-exome or genome sequencing with homozygosity mapping, we identified the homozygous c.2938G>A (p.Gly980Arg) missense mutation within the gryzun domain of TRAPPC11 in the Syrian LGMD family and the homozygous c.1287+5G>A splice-site mutation resulting in a 58 amino acid in-frame deletion (p.Ala372_Ser429del) in the foie gras domain of TRAPPC11 in the Hutterite families. TRAPPC11 encodes a component of the multiprotein TRAPP complex involved in membrane trafficking. We demonstrate that both mutations impair the binding ability of TRAPPC11 to other TRAPP complex components and disrupt the Golgi apparatus architecture. Marker trafficking experiments for the p.Ala372_Ser429del deletion indicated normal ER-to-Golgi trafficking but dramatically delayed exit from the Golgi to the cell surface. Moreover, we observed alterations of the lysosomal membrane glycoproteins lysosome-associated membrane protein 1 (LAMP1) and LAMP2 as a consequence of TRAPPC11 dysfunction supporting a defect in the transport of secretory proteins as the underlying pathomechanism.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Rosales X.Q., Tsao C.Y. Childhood onset of limb-girdle muscular dystrophy. Pediatr. Neurol. 2012;46:13–23. - PubMed
-
- Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract. Neurol. 2009;9:314–323. - PubMed
-
- Pegoraro, E., and Hoffman, E. (2000, updated 2012). Limb-Girdle Muscular Dystrophy Overview. In GeneReviews™ [Internet], Pagon RA, Bird TD, Dolan CR, et al., ed. (University of Washington, Seattle, WA, USA), 1993-. http://www.ncbi.nlm.nih.gov/books/NBK1408/ - PubMed
-
- Minetti C., Sotgia F., Bruno C., Scartezzini P., Broda P., Bado M., Masetti E., Mazzocco M., Egeo A., Donati M.A. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998;18:365–368. - PubMed
-
- Richard I., Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., Bourg N., Brenguier L., Devaud C., Pasturaud P., Roudaut C. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
