

Advanced optical imaging techniques for neurodevelopment
- PMID: 23831260
- PMCID: PMC3830703
- DOI: 10.1016/j.conb.2013.06.008
Advanced optical imaging techniques for neurodevelopment
Abstract
Over the past decade, developmental neuroscience has been transformed by the widespread application of confocal and two-photon fluorescence microscopy. Even greater progress is imminent, as recent innovations in microscopy now enable imaging with increased depth, speed, and spatial resolution; reduced phototoxicity; and in some cases without external fluorescent probes. We discuss these new techniques and emphasize their dramatic impact on neurobiology, including the ability to image neurons at depths exceeding 1mm, to observe neurodevelopment noninvasively throughout embryogenesis, and to visualize neuronal processes or structures that were previously too small or too difficult to target with conventional microscopy.
Published by Elsevier Ltd.
Figures
References
-
- Kobat D, Horton NG, Xu C. In vivo two-photon microscopy to 1.6-mm depth in mouse cortex. J Biomed Opt. 2011;16(10):106014. - PubMed
-
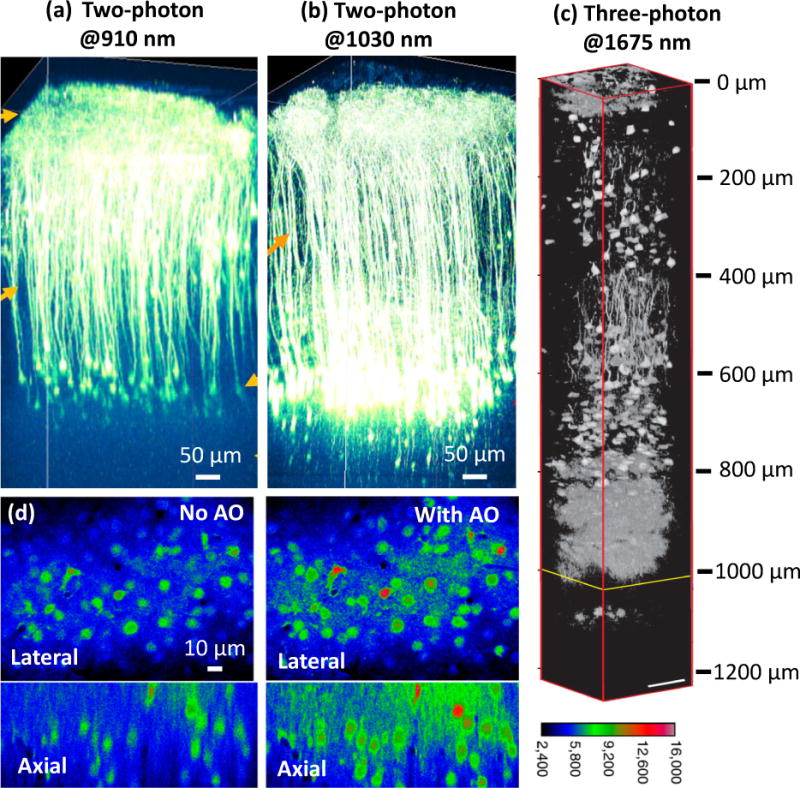
- Horton Nicholas G, Demirhan Kobat KW, Clar Catharine G, Wise Frank W, Schaffer Chris B, Xu Chris. In vivo three-photon microscopy of subcortical structures within an intact mouse brain. Nature Photonics. 2013;7:205–209. Three-photon fluorescence microscopy with a high-pulse-energy laser at ~1650 nm has enabled the in vivo high-resolution imaging of subcortical structures within an intact mouse brain with a penetration depth of ~1.4 mm. - PMC - PubMed
-
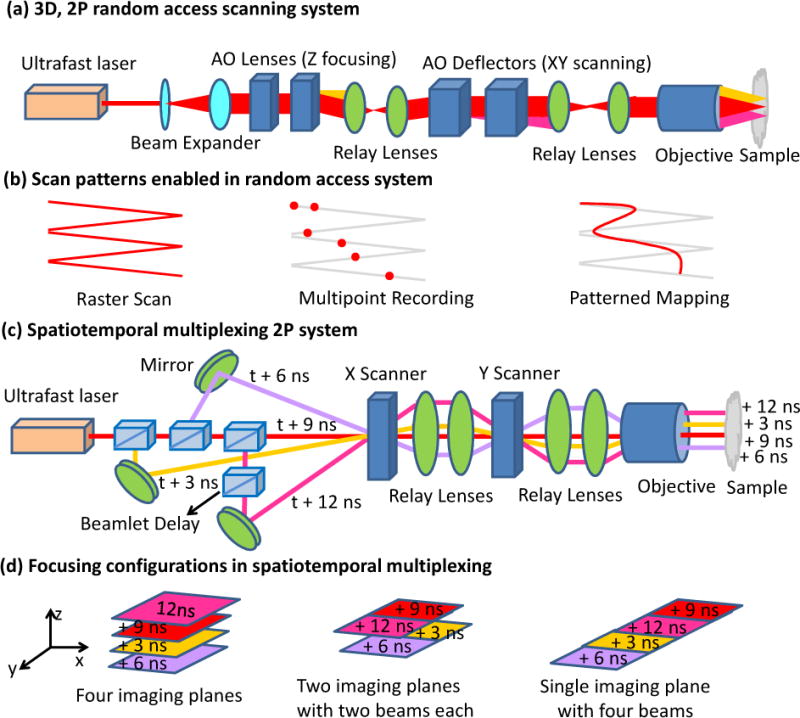
- Wang C, Ji N. Pupil-segmentation-based adaptive optical correction of a high-numerical-aperture gradient refractive index lens for two-photon fluorescence endoscopy. Opt Lett. 2012;37(11):2001–3. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
