

Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: implications for atherosclerotic plaque vulnerability
- PMID: 23832324
- PMCID: PMC3828809
- DOI: 10.1161/JAHA.113.000096
Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: implications for atherosclerotic plaque vulnerability
Abstract
Background: Vascular smooth muscle cell (VSMC) senescence and apoptosis are involved in atherosclerotic plaque vulnerability. Arginase-II (Arg-II) has been shown to promote vascular dysfunction and plaque vulnerability phenotypes in mice through uncoupling of endothelial nitric oxide synthase and activation of macrophage inflammation. The function of Arg-II in VSMCs with respect to plaque vulnerability is unknown. This study investigated the functions of Arg-II in VSMCs linking to plaque vulnerability.
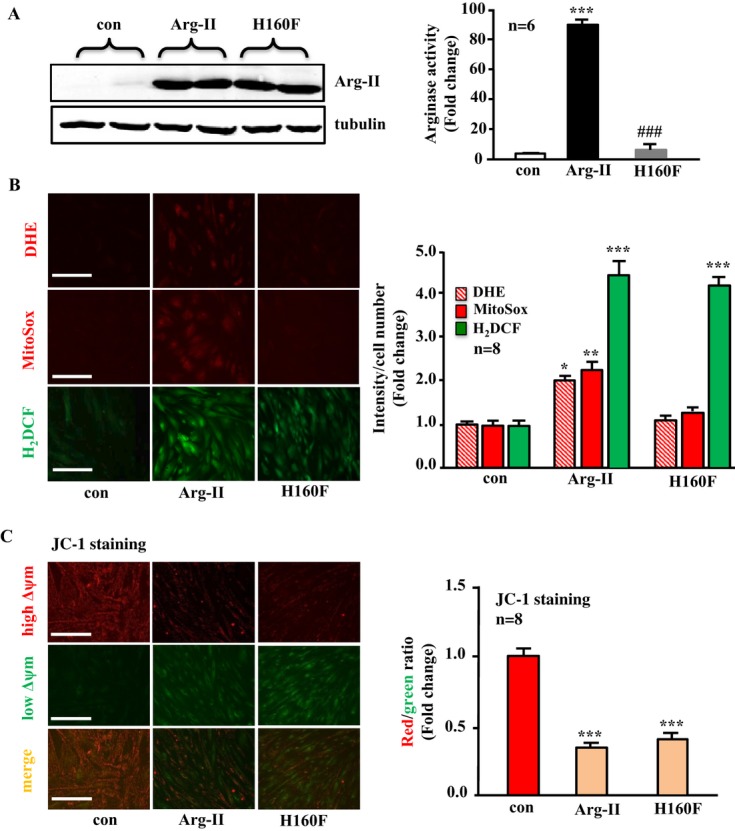
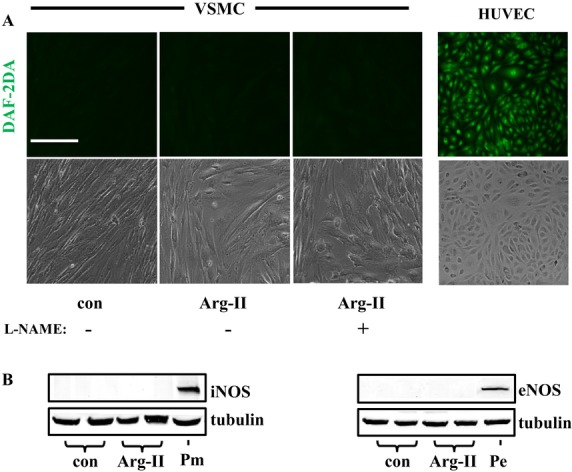
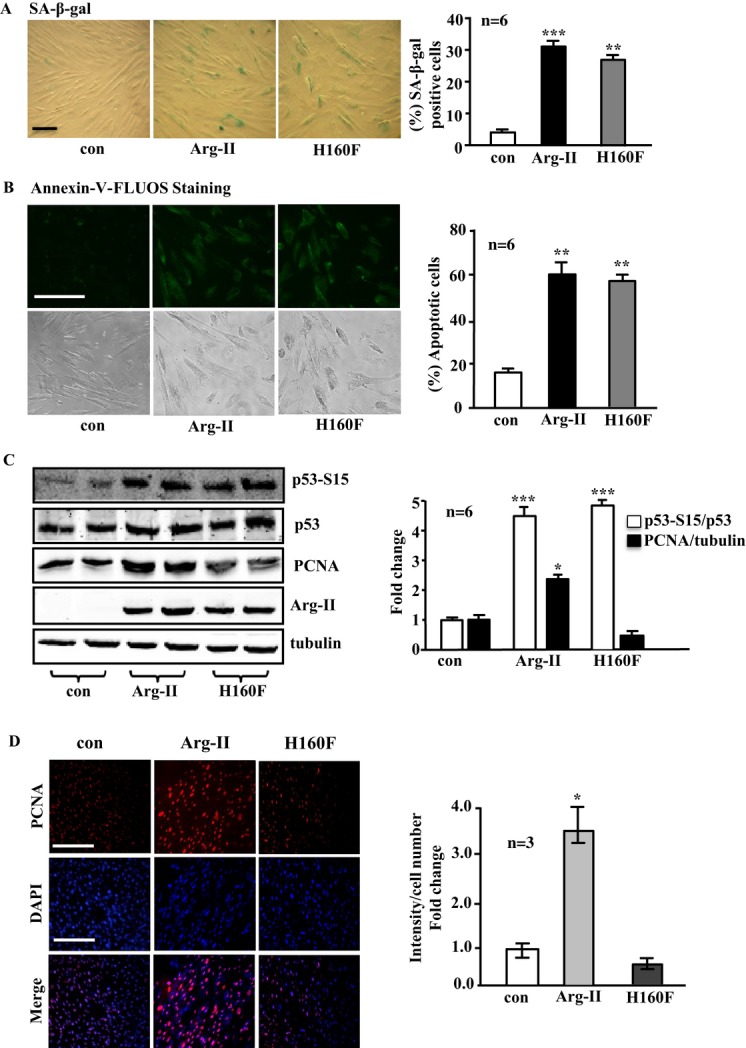
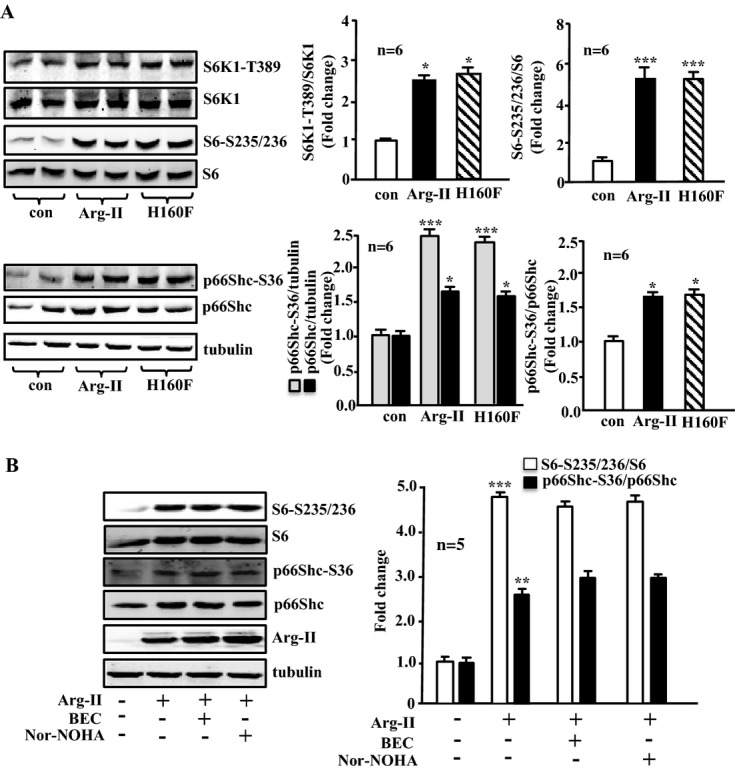
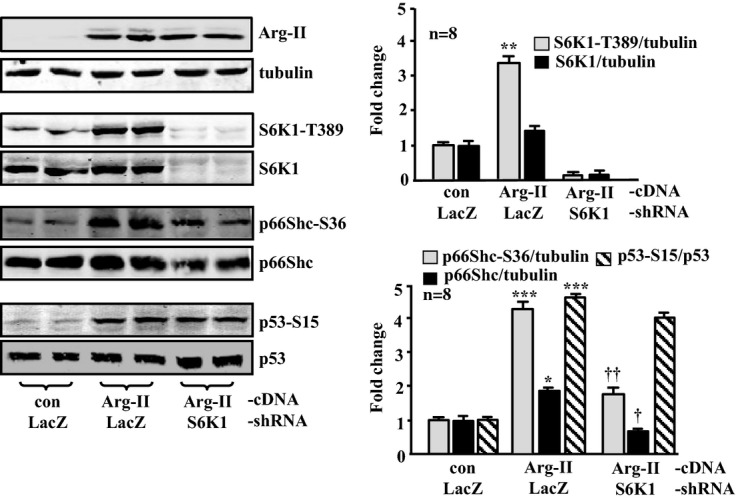
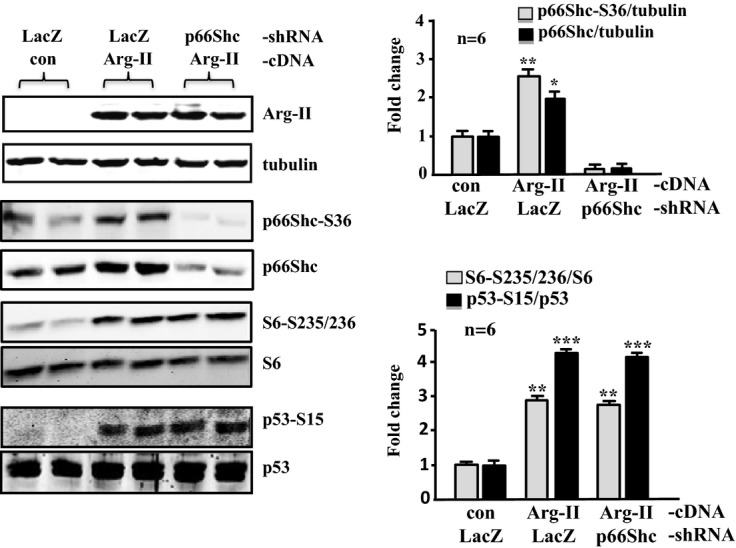
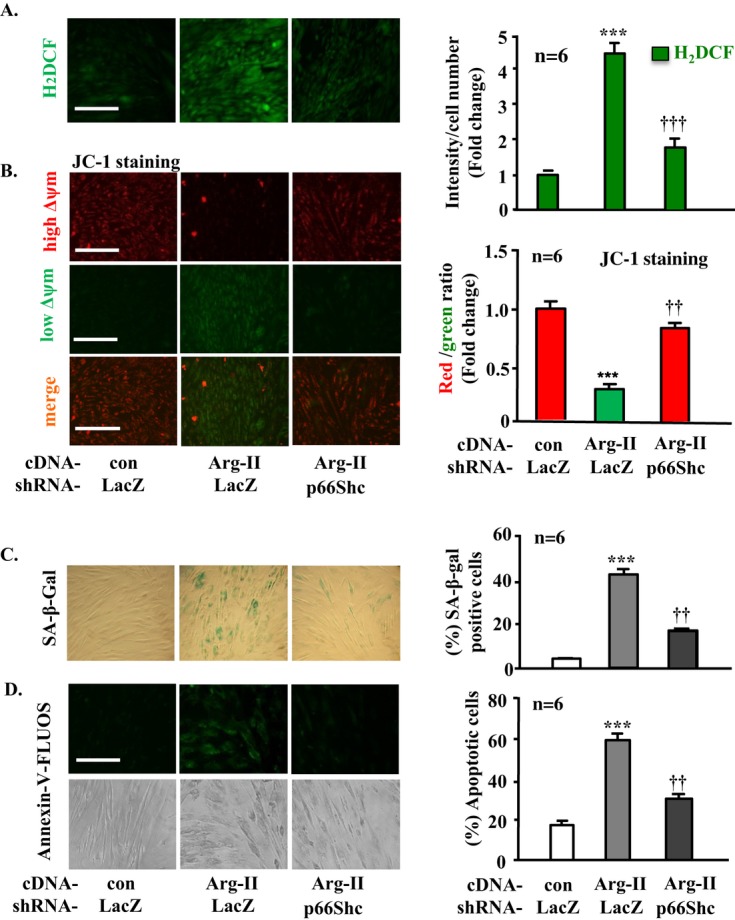
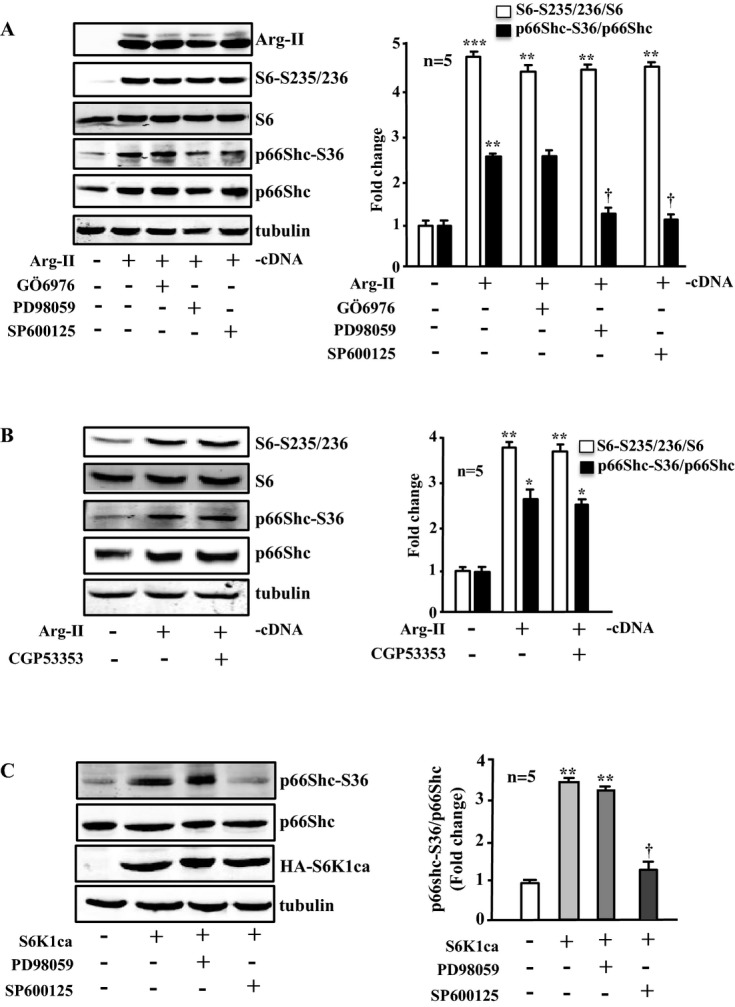
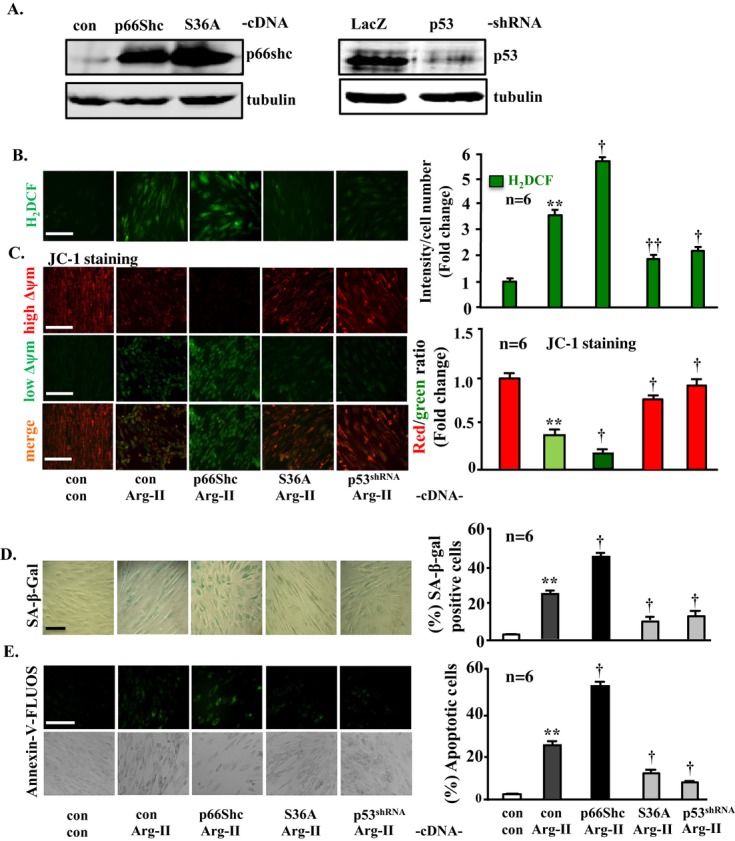
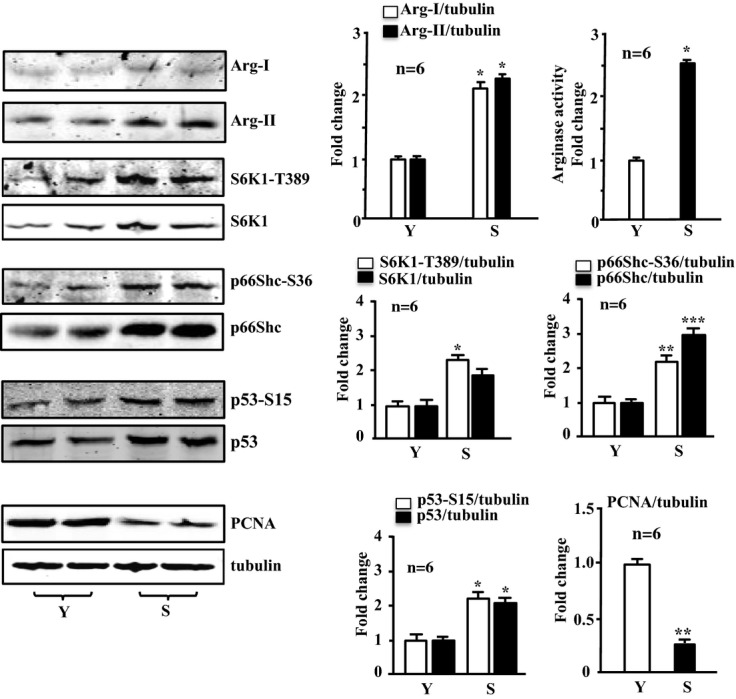
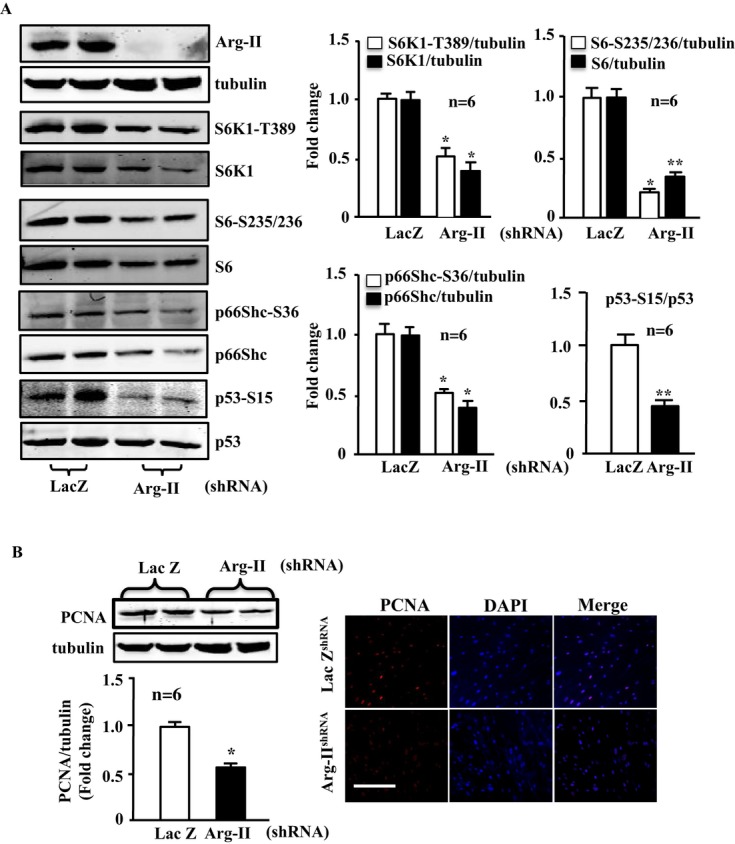
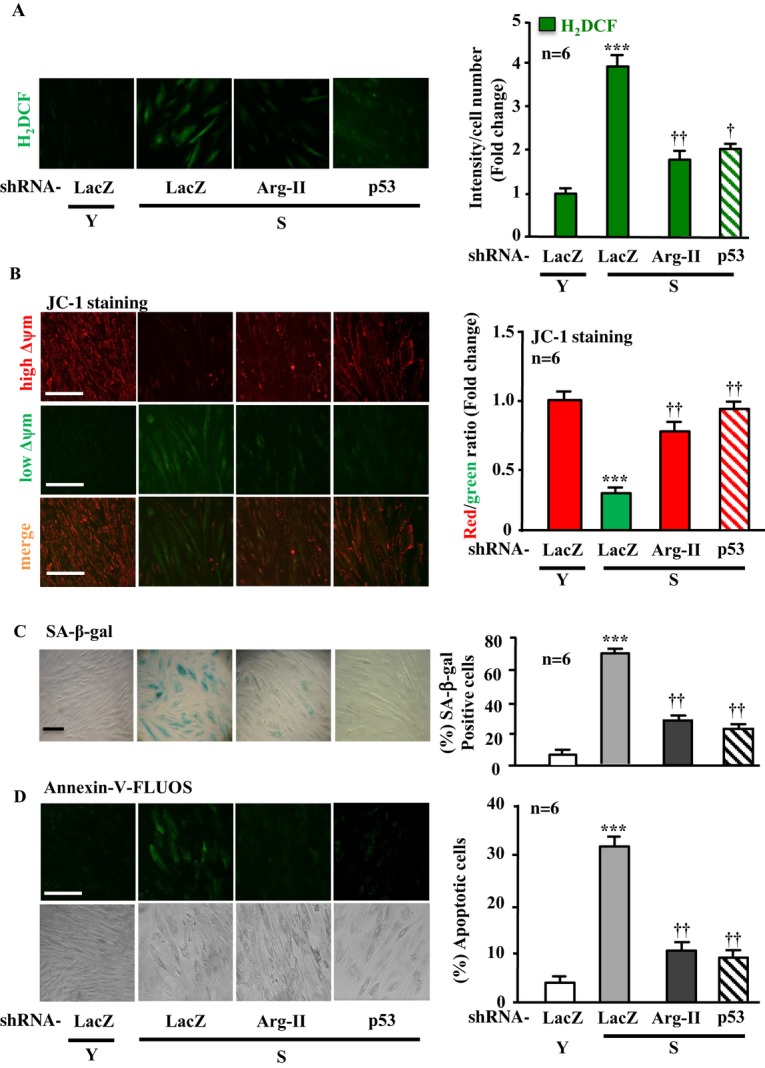
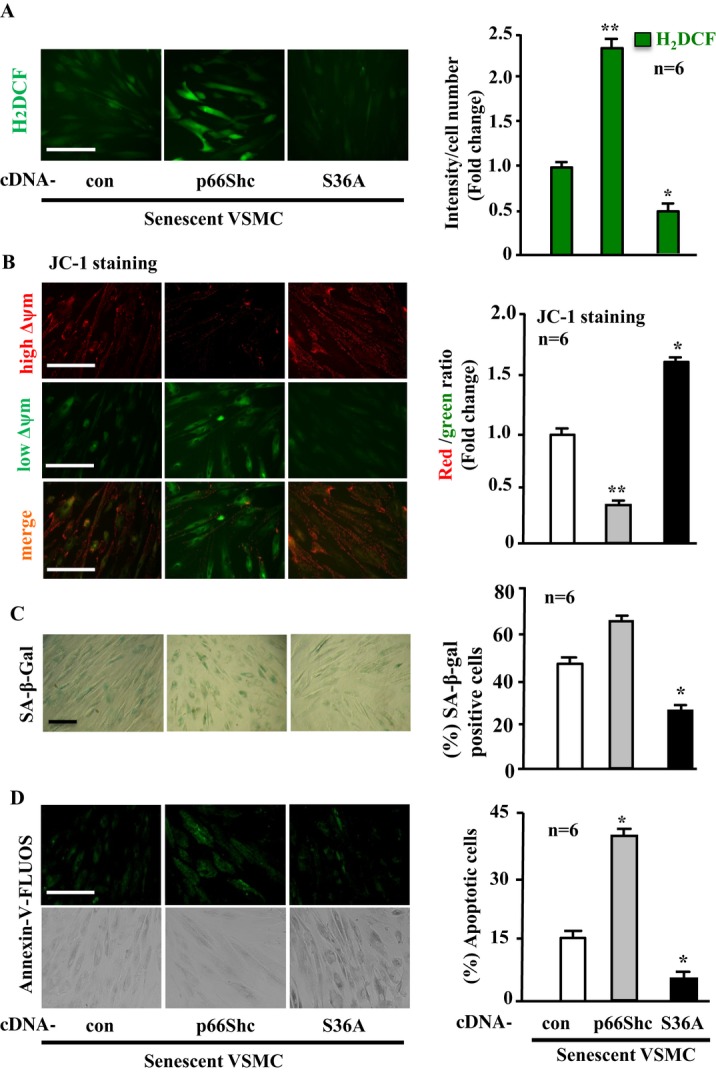
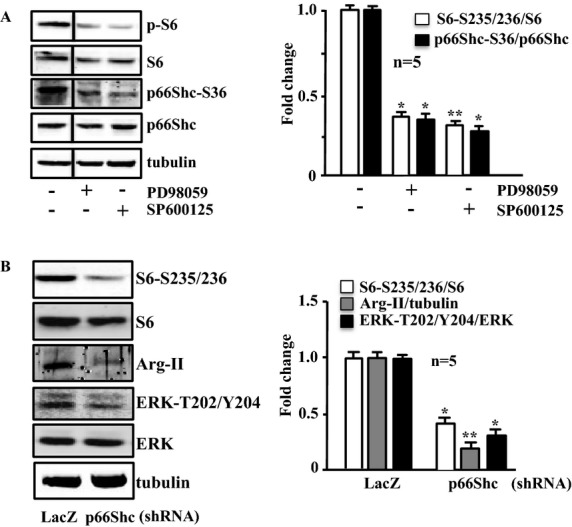
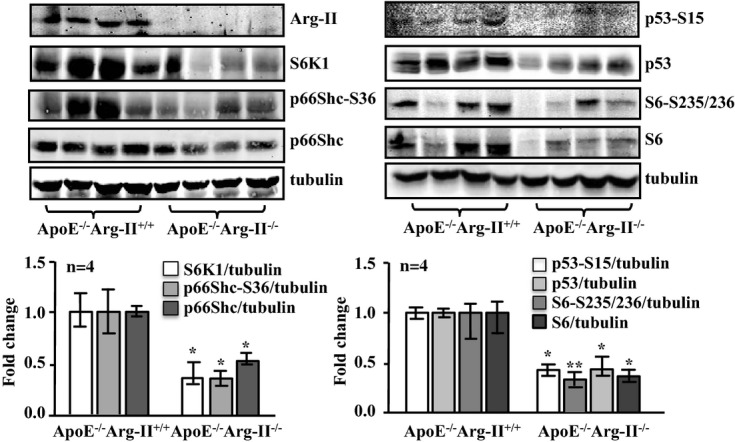
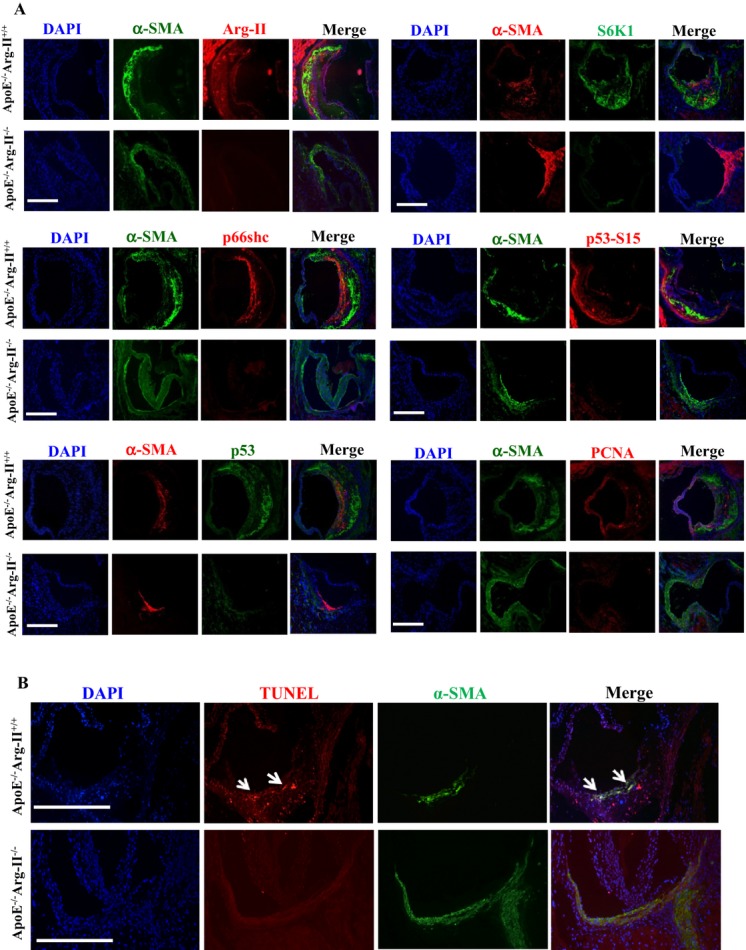
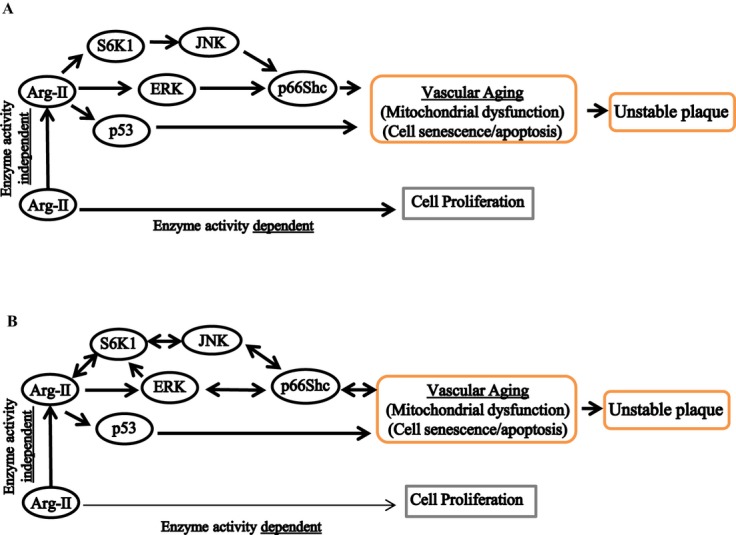
Methods and results: In vitro studies were performed on VSMCs isolated from human umbilical veins, whereas in vivo studies were performed on atherosclerosis-prone apolipoprotein E-deficient (ApoE(-/-)) mice. In nonsenescent VSMCs, overexpressing wild-type Arg-II or an l-arginine ureahydrolase inactive Arg-II mutant (H160F) caused similar effects on mitochondrial dysfunction, cell apoptosis, and senescence, which were abrogated by silencing p66Shc or p53. The activation of p66Shc but not p53 by Arg-II was dependent on extracellular signal-regulated kinases (ERKs) and sequential activation of 40S ribosomal protein S6 kinase 1 (S6K1)-c-Jun N-terminal kinases (JNKs). In senescent VSMCs, Arg-II and S6K1, ERK-p66Shc, and p53 signaling levels were increased. Silencing Arg-II reduced all these signalings and cell senescence/apoptosis. Conversely, silencing p66Shc reduced ERK and S6K1 signaling and Arg-II levels and cell senescence/apoptosis. Furthermore, genetic ablation of Arg-II in ApoE(-/-) mice reduced the aforementioned signaling and apoptotic VSMCs in the plaque of aortic roots.
Conclusions: Arg-II, independently of its l-arginine ureahydrolase activity, promotes mitochondrial dysfunction leading to VSMC senescence/apoptosis through complex positive crosstalk among S6K1-JNK, ERK, p66Shc, and p53, contributing to atherosclerotic vulnerability phenotypes in mice.
Keywords: apoptosis; arginase; p53; p66Shc; vascular smooth muscle cells.
Figures
Comment in
-
Arginase II: atherogenesis beyond enzyme activity.J Am Heart Assoc. 2013 Aug 12;2(4):e000392. doi: 10.1161/JAHA.113.000392. J Am Heart Assoc. 2013. PMID: 23938287 Free PMC article. No abstract available.
References
-
- Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002; 8:1257-1262 - PubMed
-
- Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010; 30:1282-1292 - PubMed
-
- Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012; 111:245-259 - PubMed
-
- Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006; 12:1075-1080 - PubMed
-
- Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere‐based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006; 99:156-164 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
