

The clinical maze of mitochondrial neurology
- PMID: 23835535
- PMCID: PMC3959773
- DOI: 10.1038/nrneurol.2013.126
The clinical maze of mitochondrial neurology
Abstract
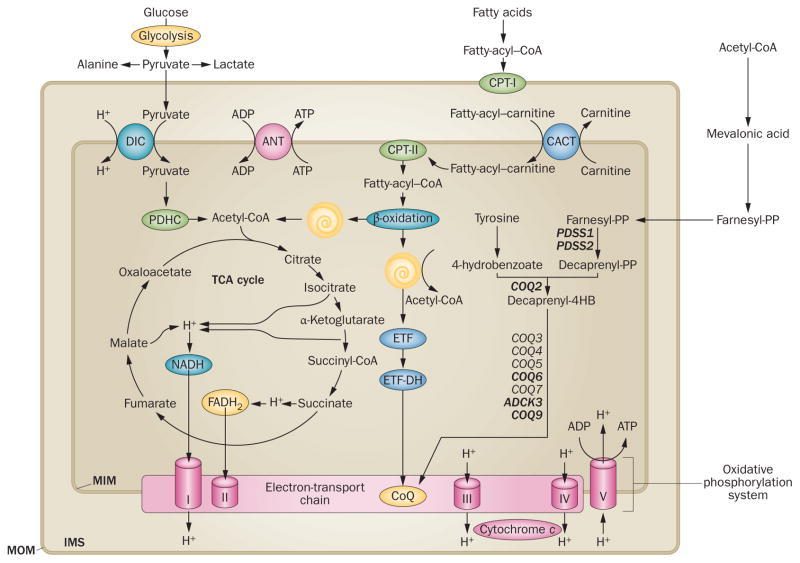
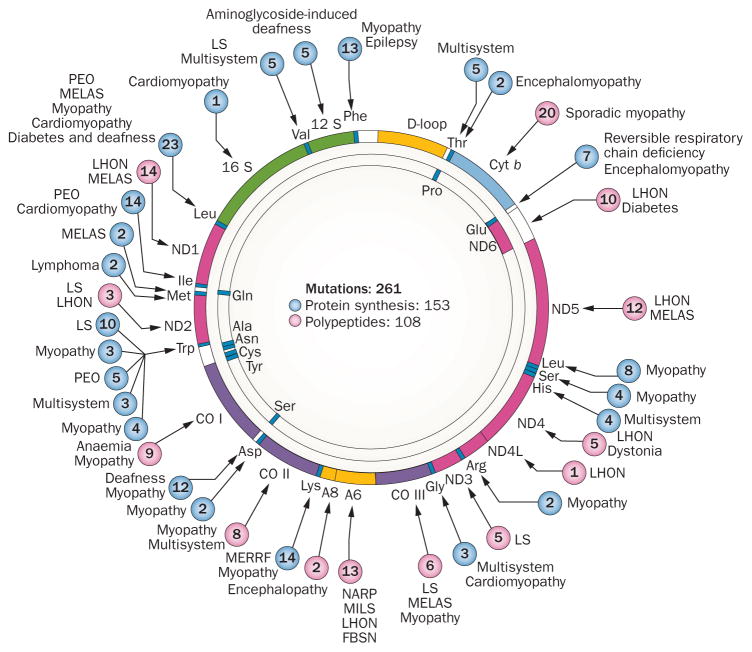
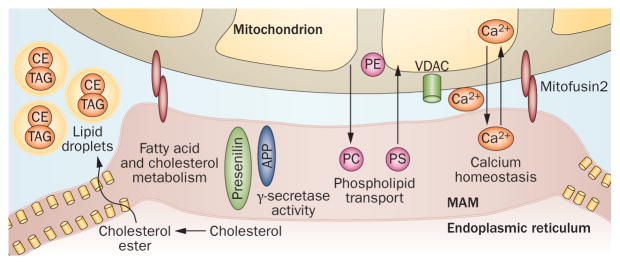
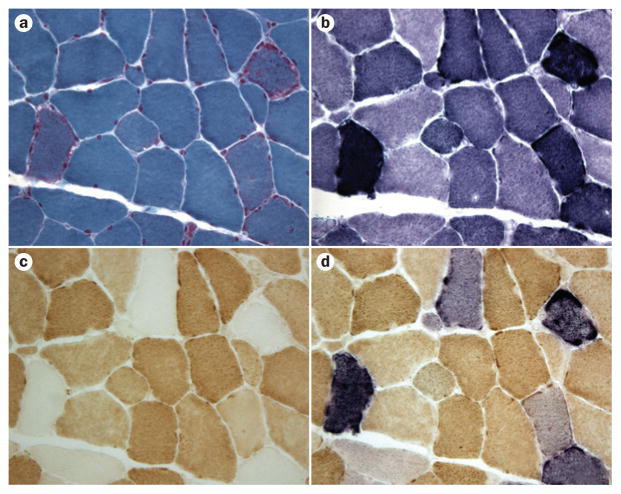
Mitochondrial diseases involve the respiratory chain, which is under the dual control of nuclear and mitochondrial DNA (mtDNA). The complexity of mitochondrial genetics provides one explanation for the clinical heterogeneity of mitochondrial diseases, but our understanding of disease pathogenesis remains limited. Classification of Mendelian mitochondrial encephalomyopathies has been laborious, but whole-exome sequencing studies have revealed unexpected molecular aetiologies for both typical and atypical mitochondrial disease phenotypes. Mendelian mitochondrial defects can affect five components of mitochondrial biology: subunits of respiratory chain complexes (direct hits); mitochondrial assembly proteins; mtDNA translation; phospholipid composition of the inner mitochondrial membrane; or mitochondrial dynamics. A sixth category-defects of mtDNA maintenance-combines features of Mendelian and mitochondrial genetics. Genetic defects in mitochondrial dynamics are especially important in neurology as they cause optic atrophy, hereditary spastic paraplegia, and Charcot-Marie-Tooth disease. Therapy is inadequate and mostly palliative, but promising new avenues are being identified. Here, we review current knowledge on the genetics and pathogenesis of the six categories of mitochondrial disorders outlined above, focusing on their salient clinical manifestations and highlighting novel clinical entities. An outline of diagnostic clues for the various forms of mitochondrial disease, as well as potential therapeutic strategies, is also discussed.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Shapira Y, Harel S, Russell A. Mitochondrial encephalomyopathies: a group of neuromuscular disorders with defects in oxidative metabolism. Isr J Med Sci. 1977;13:161–164. - PubMed
-
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. - PubMed
-
- Wallace DC, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. - PubMed
