

Metasignatures identify two major subtypes of breast cancer
- PMID: 23836026
- PMCID: PMC3615534
- DOI: 10.1038/psp.2013.11
Metasignatures identify two major subtypes of breast cancer
Abstract
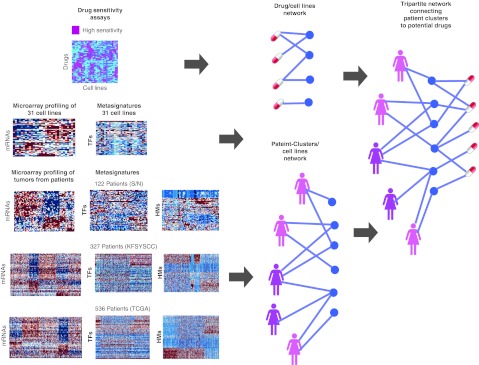
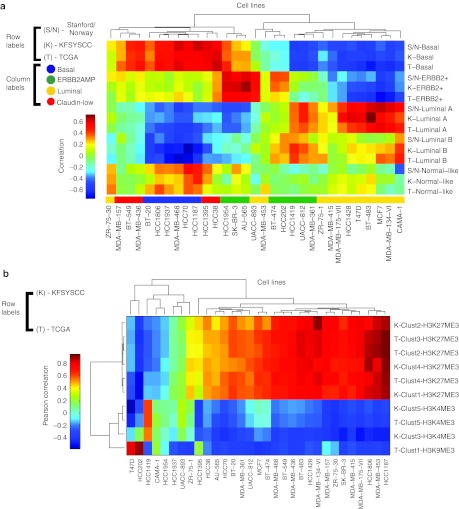
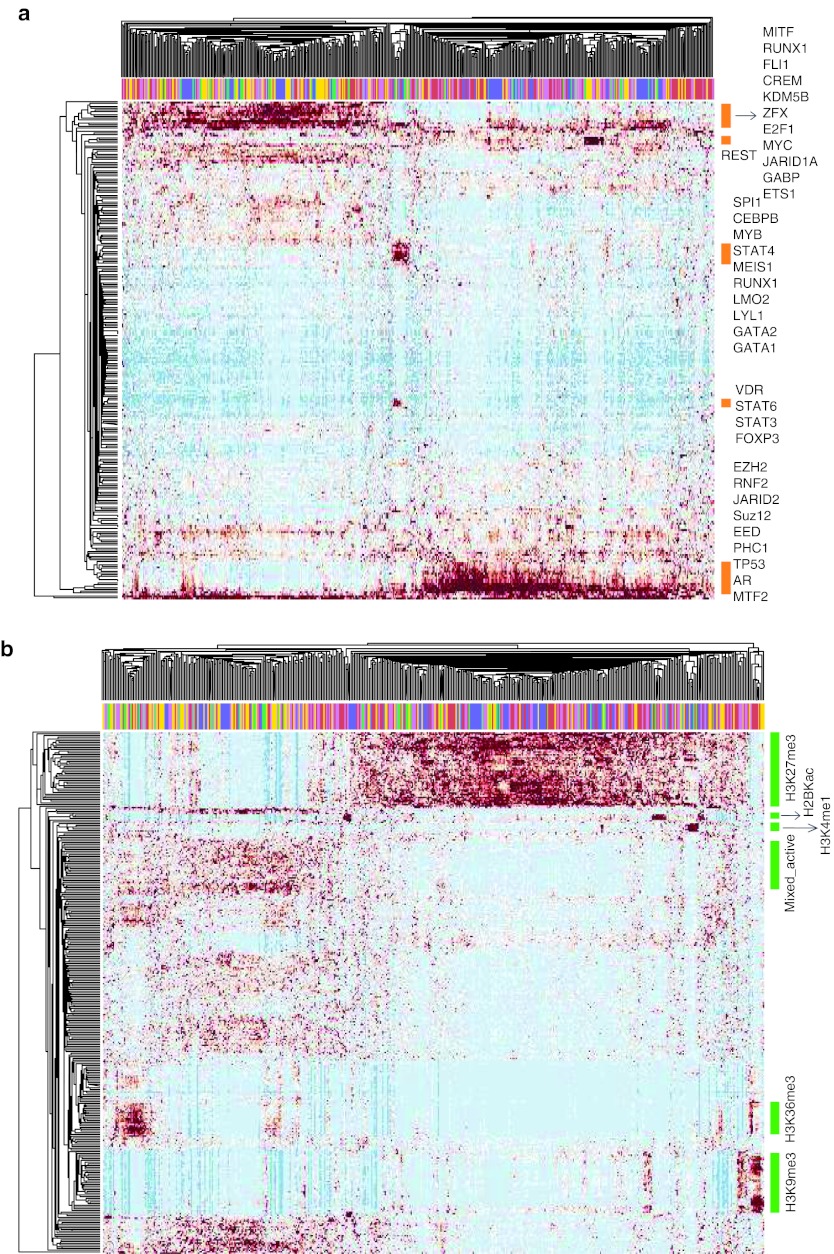
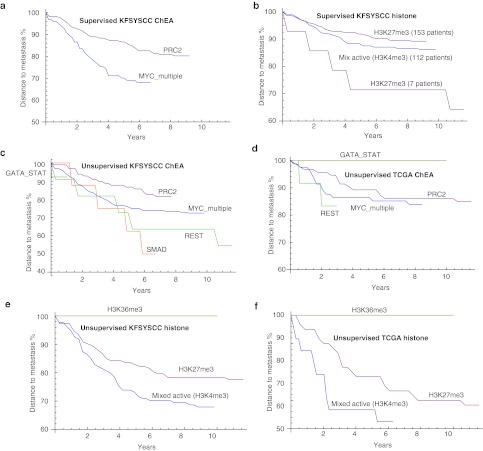
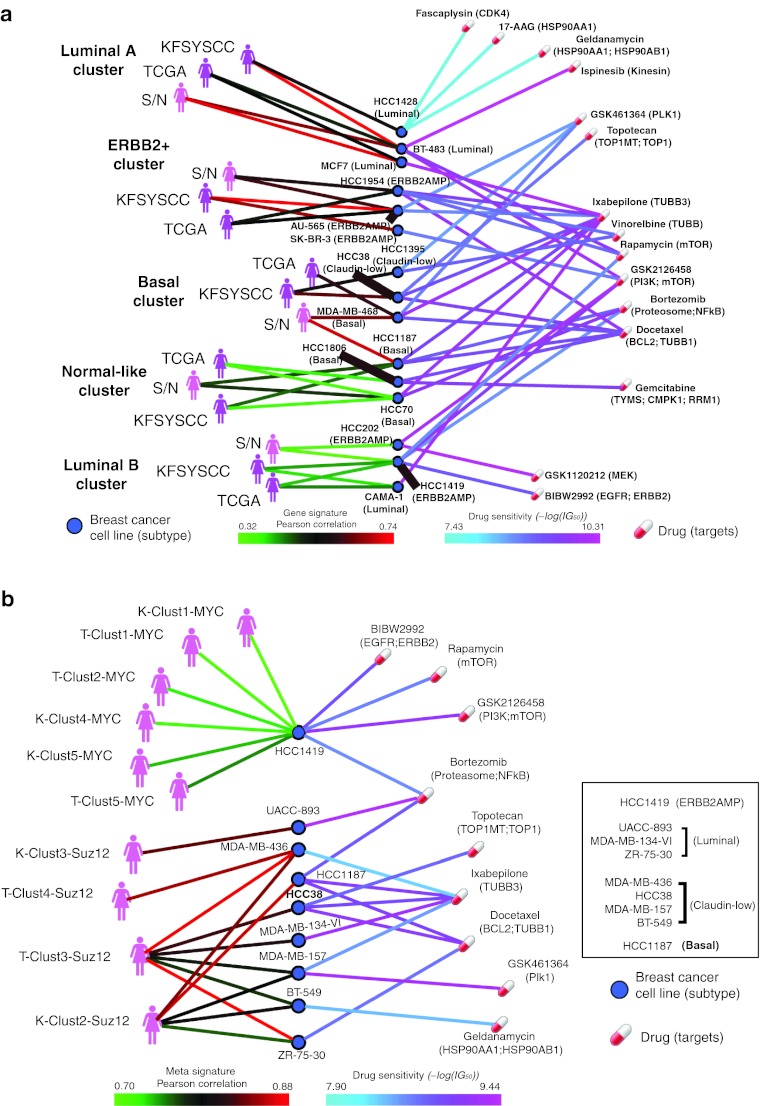
Genome-wide expression data from tumors and cell lines in breast cancer, together with drug response of cell lines, open prospects for integrative analyses that can lead to better personalized therapy. Drug responses and expression data collected from cell lines and tumors were used to generate tripartite networks connecting clusters of patients to cell lines and cell lines to drugs, to connect drugs to patients. Various approaches were applied to connect cell lines to tumor clusters: a standard method that uses a biomarker gene set, and new methods that compute metasignatures for transcription factors and histone modifications given upregulated genes in cell lines or tumors. The results from the metasignature analysis identify two major clusters of patients: one enriched for active histone marks and one for repressive marks. The tumors enriched for activation marks are correlated with poor prognosis. Overall, the analyses suggest new patient clustering, discover dysregulated pathways, and recommend individualized use of drugs to treat subsets of patients.CPT: Pharmacometrics & Systems Pharmacology (2013) 2, e35; doi:10.1038/psp.2013.11; advance online publication 27 March 2013.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
