

Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile
- PMID: 23836560
- PMCID: PMC3744994
- DOI: 10.1182/blood-2013-05-501072
Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile
Abstract
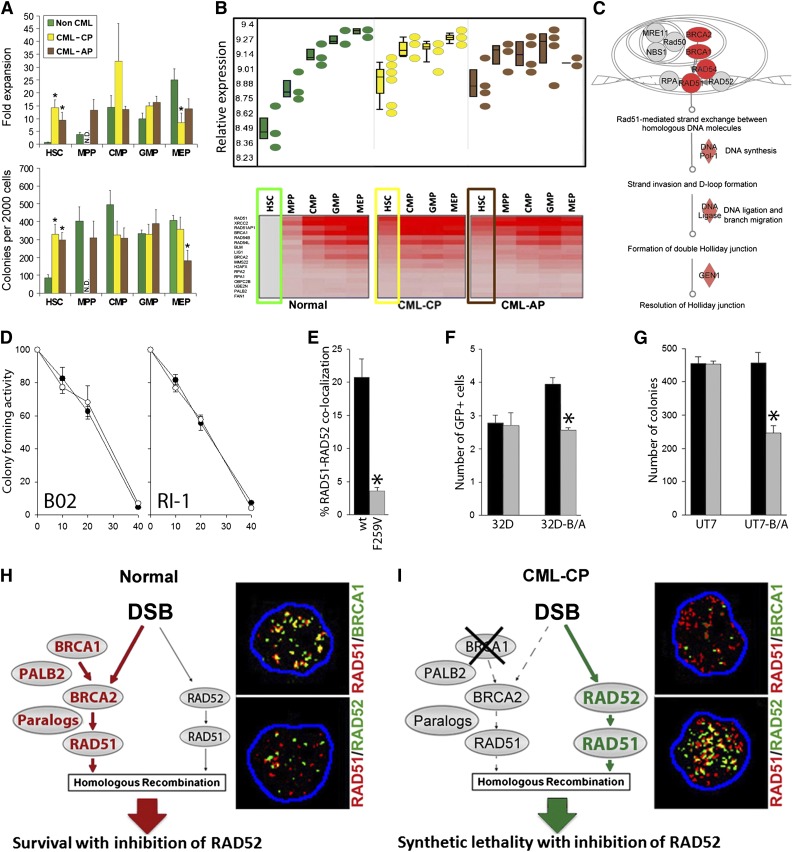
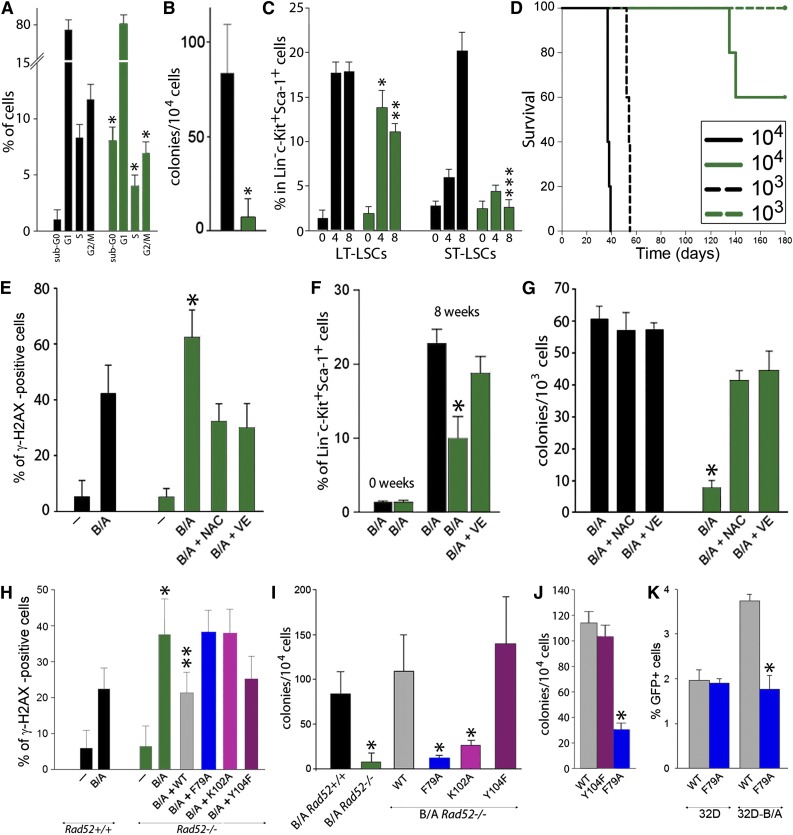
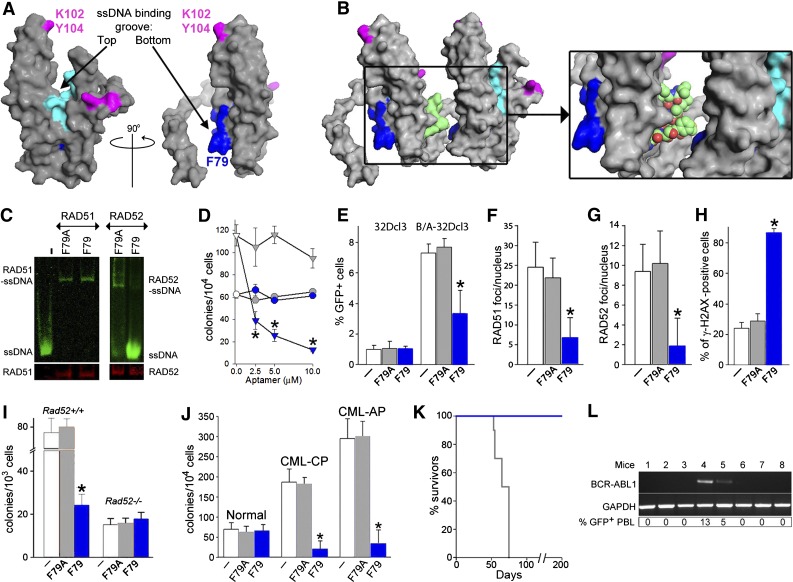
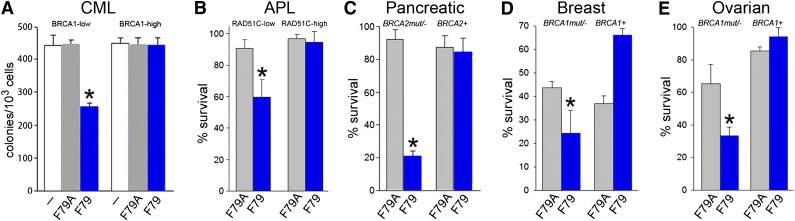
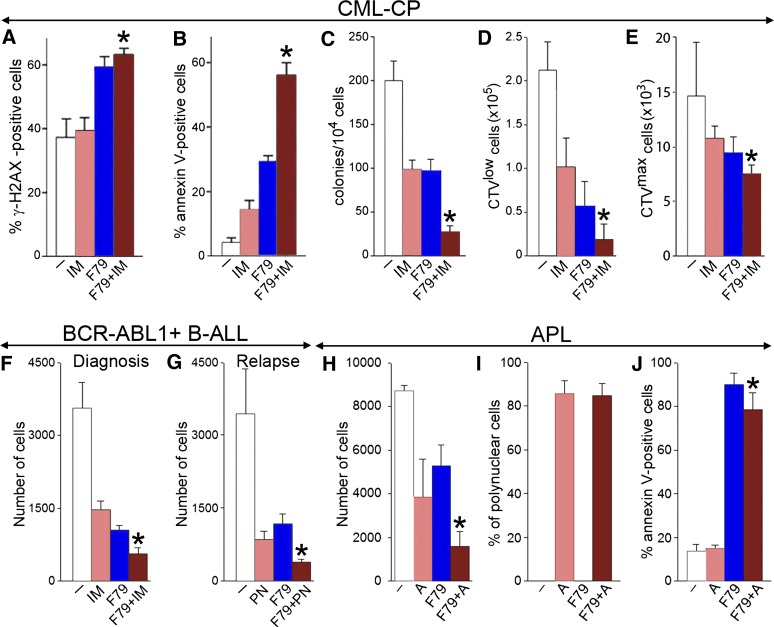
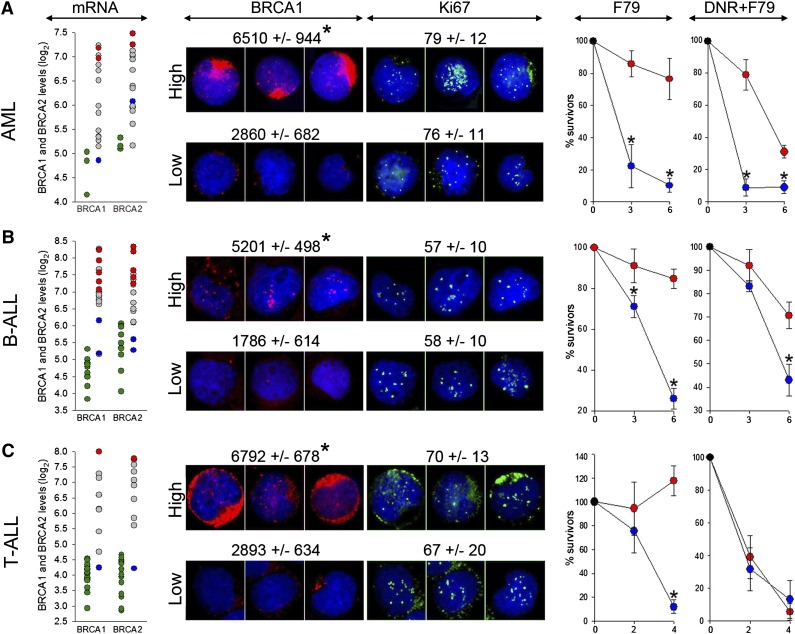
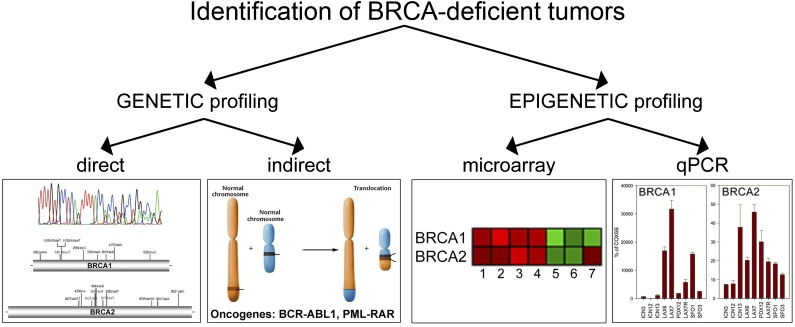
Homologous recombination repair (HRR) protects cells from the lethal effect of spontaneous and therapy-induced DNA double-stand breaks. HRR usually depends on BRCA1/2-RAD51, and RAD52-RAD51 serves as back-up. To target HRR in tumor cells, a phenomenon called "synthetic lethality" was applied, which relies on the addiction of cancer cells to a single DNA repair pathway, whereas normal cells operate 2 or more mechanisms. Using mutagenesis and a peptide aptamer approach, we pinpointed phenylalanine 79 in RAD52 DNA binding domain I (RAD52-phenylalanine 79 [F79]) as a valid target to induce synthetic lethality in BRCA1- and/or BRCA2-deficient leukemias and carcinomas without affecting normal cells and tissues. Targeting RAD52-F79 disrupts the RAD52-DNA interaction, resulting in the accumulation of toxic DNA double-stand breaks in malignant cells, but not in normal counterparts. In addition, abrogation of RAD52-DNA interaction enhanced the antileukemia effect of already-approved drugs. BRCA-deficient status predisposing to RAD52-dependent synthetic lethality could be predicted by genetic abnormalities such as oncogenes BCR-ABL1 and PML-RAR, mutations in BRCA1 and/or BRCA2 genes, and gene expression profiles identifying leukemias displaying low levels of BRCA1 and/or BRCA2. We believe this work may initiate a personalized therapeutic approach in numerous patients with tumors displaying encoded and functional BRCA deficiency.
Figures
References
-
- Frame FM, Maitland NJ. Cancer stem cells, models of study and implications of therapy resistance mechanisms. Adv Exp Med Biol. 2011;720:105–118. - PubMed
-
- Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270(1):1–9. - PubMed
-
- Callén E, Jankovic M, Difilippantonio S, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130(1):63–75. - PubMed
-
- Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–817. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
- R21 CA133646/CA/NCI NIH HHS/United States
- P01CA70970/CA/NCI NIH HHS/United States
- 1R01CA134458/CA/NCI NIH HHS/United States
- P01 CA070970/CA/NCI NIH HHS/United States
- CAF/08/09/CSO_/Chief Scientist Office/United Kingdom
- R01 CA157644/CA/NCI NIH HHS/United States
- 1R21CA133646/CA/NCI NIH HHS/United States
- SCD/04/CSO_/Chief Scientist Office/United Kingdom
- R01 CA172558/CA/NCI NIH HHS/United States
- R01 CA169458/CA/NCI NIH HHS/United States
- T32 CA078586/CA/NCI NIH HHS/United States
- R01 CA139032/CA/NCI NIH HHS/United States
- R01CA157644/CA/NCI NIH HHS/United States
- CRUK_/Cancer Research UK/United Kingdom
- R01CA139032/CA/NCI NIH HHS/United States
- R01 CA134458/CA/NCI NIH HHS/United States
- R01 CA137060/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
