

Rapid evolution of the human gut virome
- PMID: 23836644
- PMCID: PMC3725073
- DOI: 10.1073/pnas.1300833110
Rapid evolution of the human gut virome
Abstract
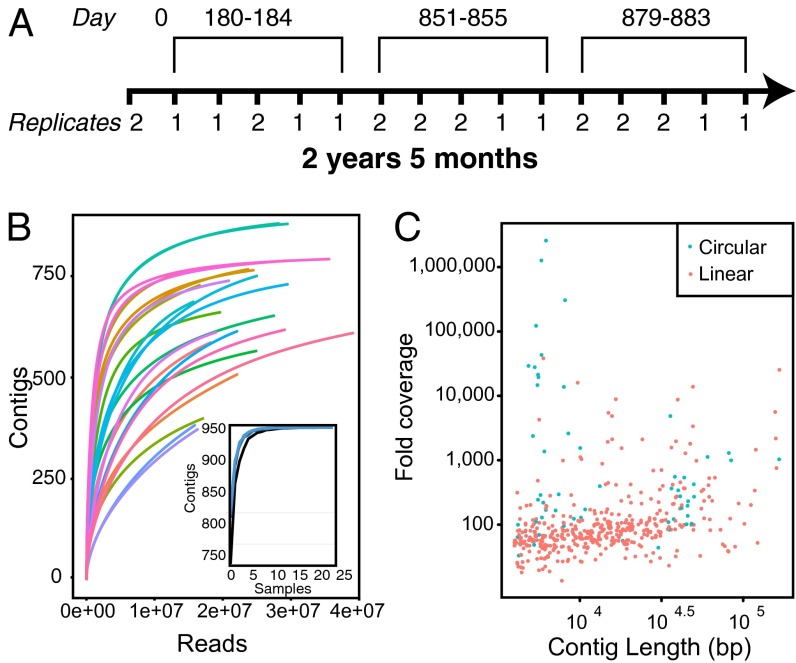
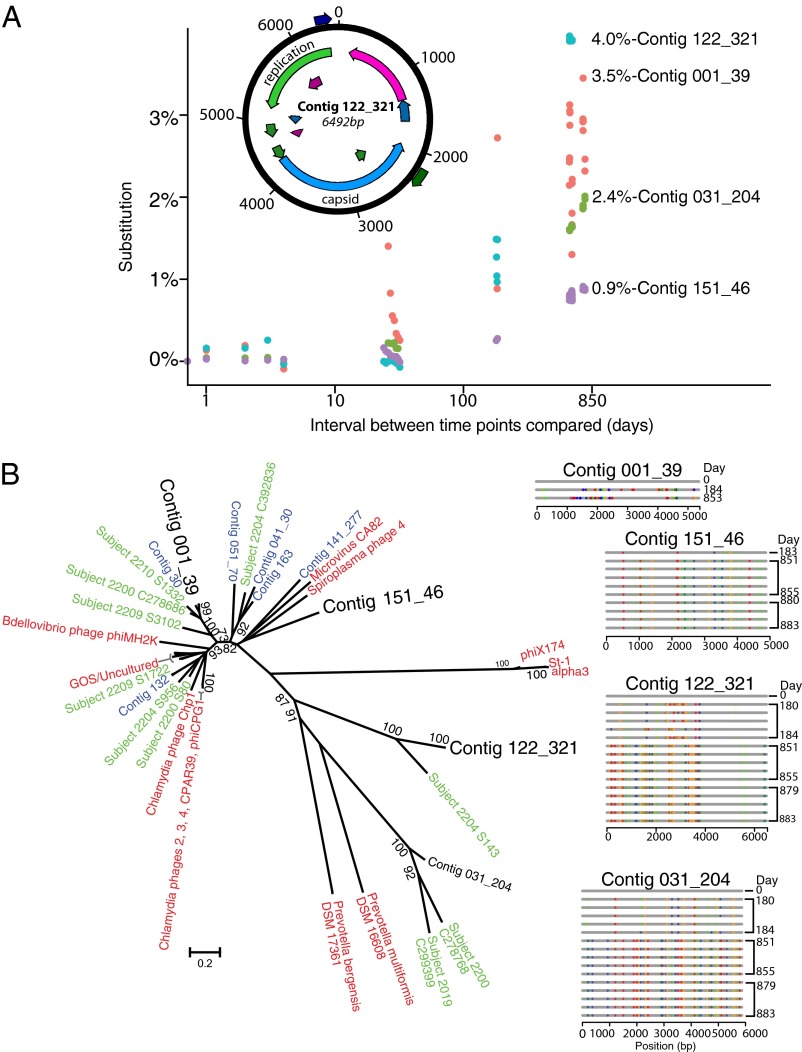
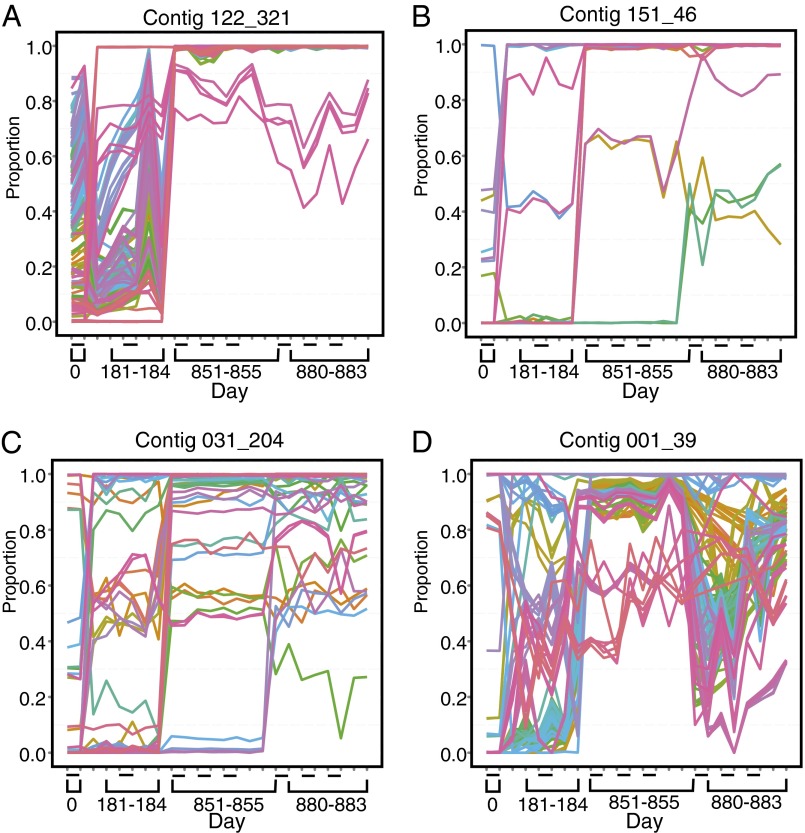
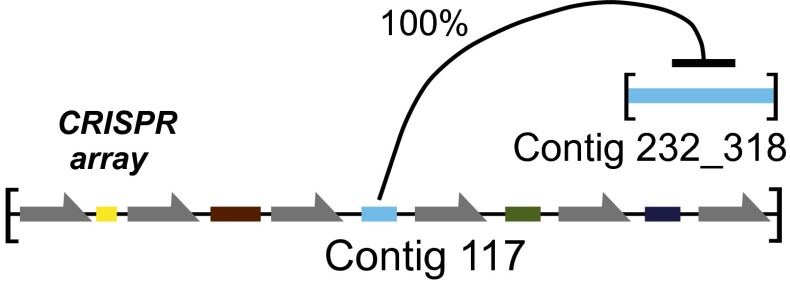
Humans are colonized by immense populations of viruses, which metagenomic analysis shows are mostly unique to each individual. To investigate the origin and evolution of the human gut virome, we analyzed the viral community of one adult individual over 2.5 y by extremely deep metagenomic sequencing (56 billion bases of purified viral sequence from 24 longitudinal fecal samples). After assembly, 478 well-determined contigs could be identified, which are inferred to correspond mostly to previously unstudied bacteriophage genomes. Fully 80% of these types persisted throughout the duration of the 2.5-y study, indicating long-term global stability. Mechanisms of base substitution, rates of accumulation, and the amount of variation varied among viral types. Temperate phages showed relatively lower mutation rates, consistent with replication by accurate bacterial DNA polymerases in the integrated prophage state. In contrast, Microviridae, which are lytic bacteriophages with single-stranded circular DNA genomes, showed high substitution rates (>10(-5) per nucleotide each day), so that sequence divergence over the 2.5-y period studied approached values sufficient to distinguish new viral species. Longitudinal changes also were associated with diversity-generating retroelements and virus-encoded Clustered Regularly Interspaced Short Palindromic Repeats arrays. We infer that the extreme interpersonal diversity of human gut viruses derives from two sources, persistence of a small portion of the global virome within the gut of each individual and rapid evolution of some long-term virome members.
Keywords: CRISPR; diversity generating retroelement; metagenomics; microbiome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Comment in
-
Viral evolution: Variation in the gut virome.Nat Rev Microbiol. 2013 Sep;11(9):596. doi: 10.1038/nrmicro3092. Epub 2013 Jul 22. Nat Rev Microbiol. 2013. PMID: 23872943 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
