

Approach to the patient: the adult with congenital adrenal hyperplasia
- PMID: 23837188
- PMCID: PMC3701266
- DOI: 10.1210/jc.2013-1440
Approach to the patient: the adult with congenital adrenal hyperplasia
Abstract
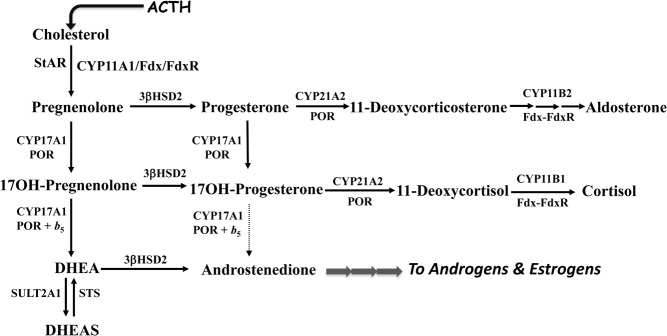
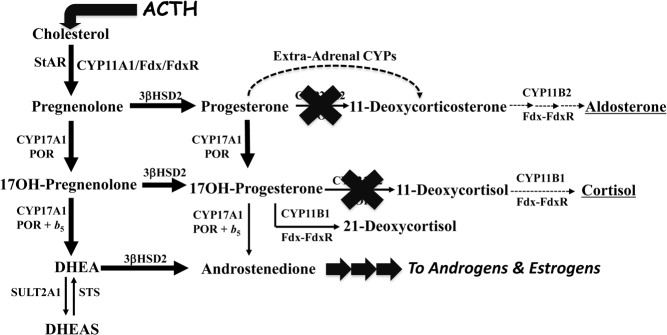
The most common form of congenital adrenal hyperplasia is steroid 21-hydroxylase deficiency (21OHD). When the nonclassical (mild) form is included, 21OHD is the most common genetic disease in human beings. With the advent of pharmaceutical preparation of glucocorticoids starting in the 1960s and newborn screening starting in the 1990s, the majority of children with 21OHD are reaching adulthood, which has yielded a cohort of patients with, in essence, a new disease. Only recently have some data emerged from cohorts of adults with 21OHD, and in some centers, experience with the management of these patients is growing. These patients suffer from poor health, infertility, characteristic tumors in the adrenal glands and gonads, and consequences of chronic glucocorticoid therapy. Their care is fragmented and inconsistent, and many stop taking their medications out of frustration. Internal medicine residents and endocrinology fellows receive little training in their care, which further discourages their seeking medical attention. Adults with 21OHD have a different physiology from patients with Addison's disease or other androgen excess states, and their needs are different than those of young children with 21OHD. Consequently, their care requires unorthodox treatment and monitoring strategies foreign to most endocrine practitioners. Our goal for this article is to review their physiology, complications, and needs in order to develop rational and effective treatment and monitoring strategies.
Figures
References
-
- Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349:776–788 - PubMed
-
- Therrell BL, Jr, Berenbaum SA, Manter-Kapanke V, et al. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–590 - PubMed
-
- Joint LWPES/ESPE CAH Working Group Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab. 2002;87:4048–4053 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
