

A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome
- PMID: 23840644
- PMCID: PMC3696103
- DOI: 10.1371/journal.pone.0067263
A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome
Abstract
Aims: With ageing extracellular material is deposited in Bruch's membrane, as drusen. Lipofuscin is deposited in retinal pigment epithelial cells. Both of these changes are associated with age related macular degeneration, a disease now believed to involve chronic inflammation at the retinal-choroidal interface. We hypothesise that these molecules may act as danger signals, causing the production of inflammatory chemokines and cytokines by the retinal pigment epithelium, via activation of pattern recognition receptors.
Methods: ARPE-19 cells were stimulated in vitro with the following reported components of drusen: amyloid-ß (1-42), Carboxyethylpyrrole (CEP) modified proteins (CEP-HSA), Nε-(Carboxymethyl)lysine (CML) modified proteins and aggregated vitronectin. The cells were also stimulated with the major fluorophore of lipofuscin: N-retinylidene-N-retinylethanolamine (A2E). Inflammatory chemokine and cytokine production was assessed using Multiplex assays and ELISA. The mechanistic evaluation of the NLRP3 inflammasome pathway was assessed in a stepwise fashion.
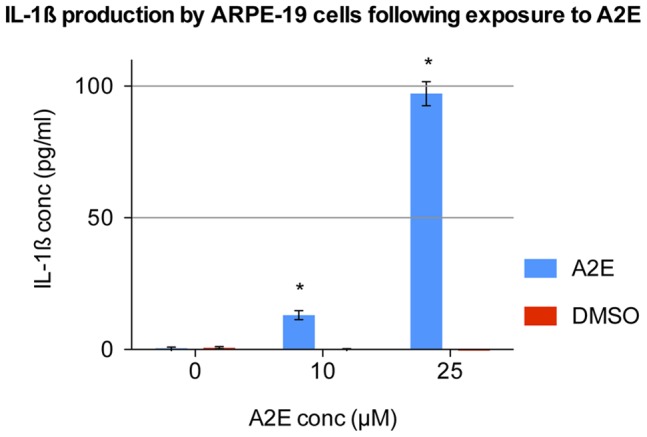
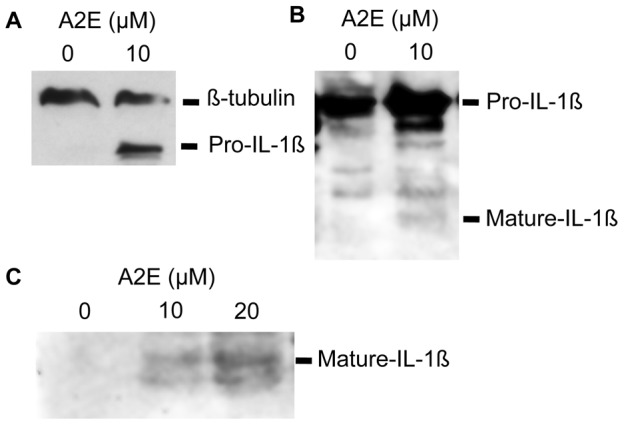
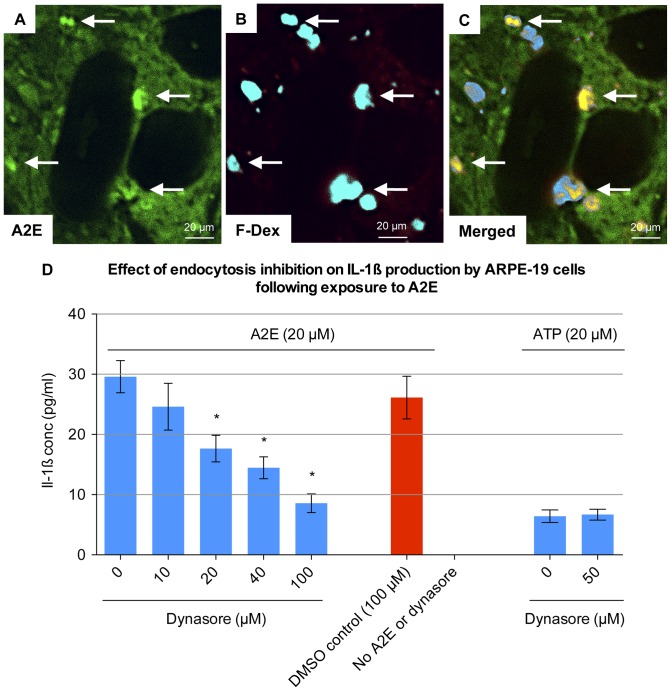
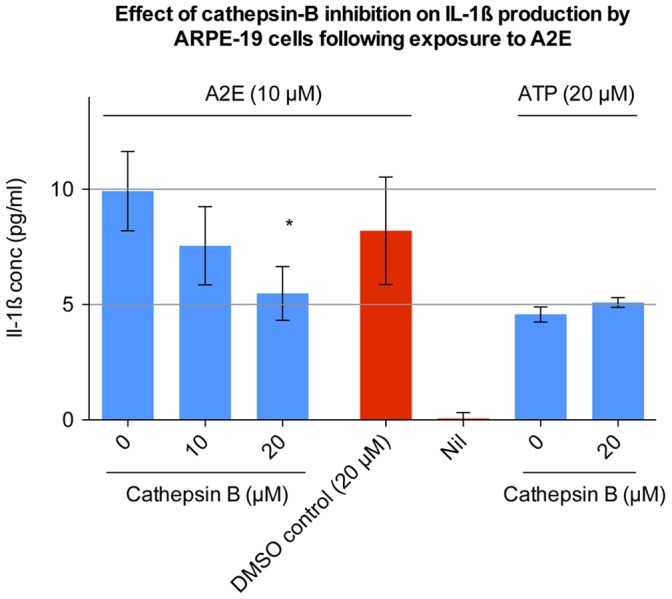
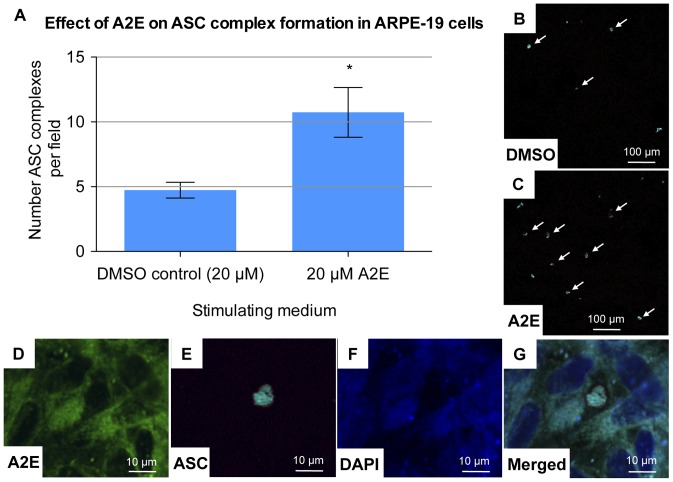
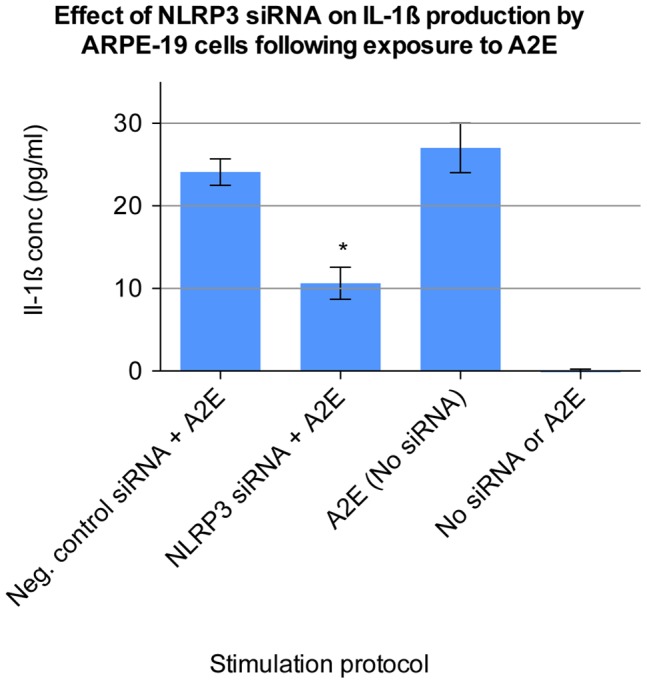
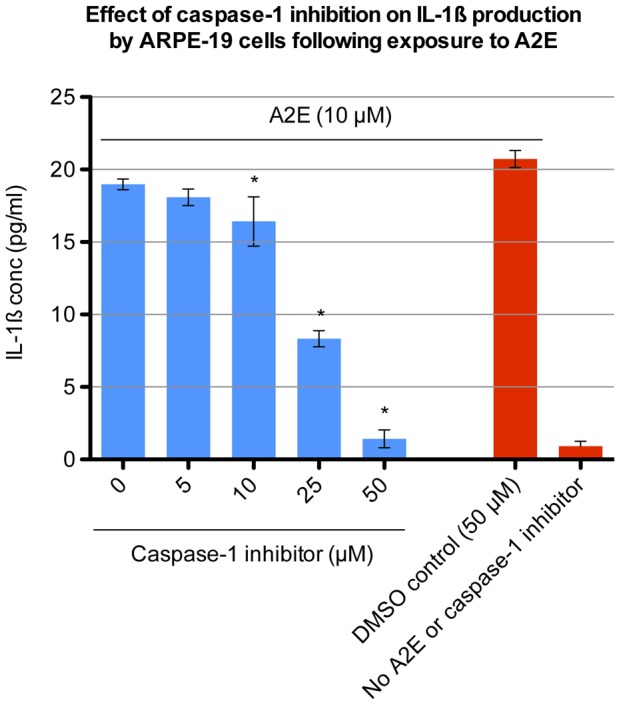
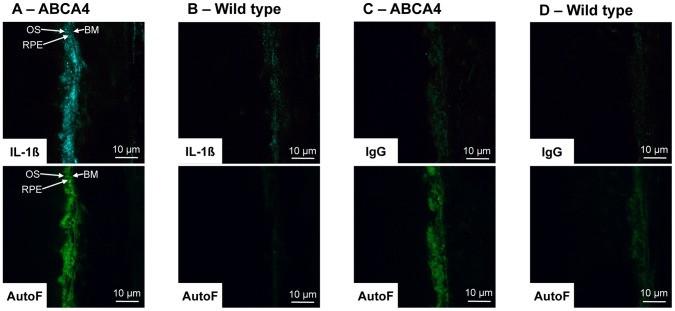
Results: Of all the molecules tested only A2E induced inflammatory chemokine and cytokine production. 25 µM A2E induced the production of significantly increased levels of the chemokines IL-8, MCP-1, MCG and MIP-1α, the cytokines IL-1ß, IL-2, IL-6, and TNF-α, and the protein VEGF-A. The release of IL-1ß was studied further, and was determined to be due to NLRP3 inflammasome activation. The pathway of activation involved endocytosis of A2E, and the three inflammasome components NLRP3, ASC and activated caspase-1. Immunohistochemical staining of ABCA4 knockout mice, which show progressive accumulation of A2E levels with age, showed increased amounts of IL-1ß proximal to the retinal pigment epithelium.
Conclusions: A2E has the ability to stimulate inflammatory chemokine and cytokine production by RPE cells. The pattern recognition receptor NLRP3 is involved in this process. This provides further evidence for the link between A2E, inflammation, and the pathogenesis of AMD. It also supports the recent discovery of NLRP3 inflammasome activation in AMD.
Conflict of interest statement
Figures
References
-
- Klein R, Klein BE, Jenson SC, Meuer SM (1997) The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology 104: 7–21. - PubMed
-
- Rahmani B, Tielsch JM, Katz J, Gottsch J, Quigley H, et al. (1996) The cause-specific prevalence of visual impairment in an urban population. The Baltimore Eye Survey. Ophthalmology 103: 1721–1726. - PubMed
-
- Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP (2003) Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol 48: 257–293. - PubMed
-
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, et al. (2006) Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 355: 1419–1431. - PubMed
-
- Feeney-Burns L, Hilderbrand ES, Eldridge S (1984) Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Investigative Ophthalmology Visual Science 25: 195–200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
