

Role of ABL family kinases in cancer: from leukaemia to solid tumours
- PMID: 23842646
- PMCID: PMC3935732
- DOI: 10.1038/nrc3563
Role of ABL family kinases in cancer: from leukaemia to solid tumours
Abstract
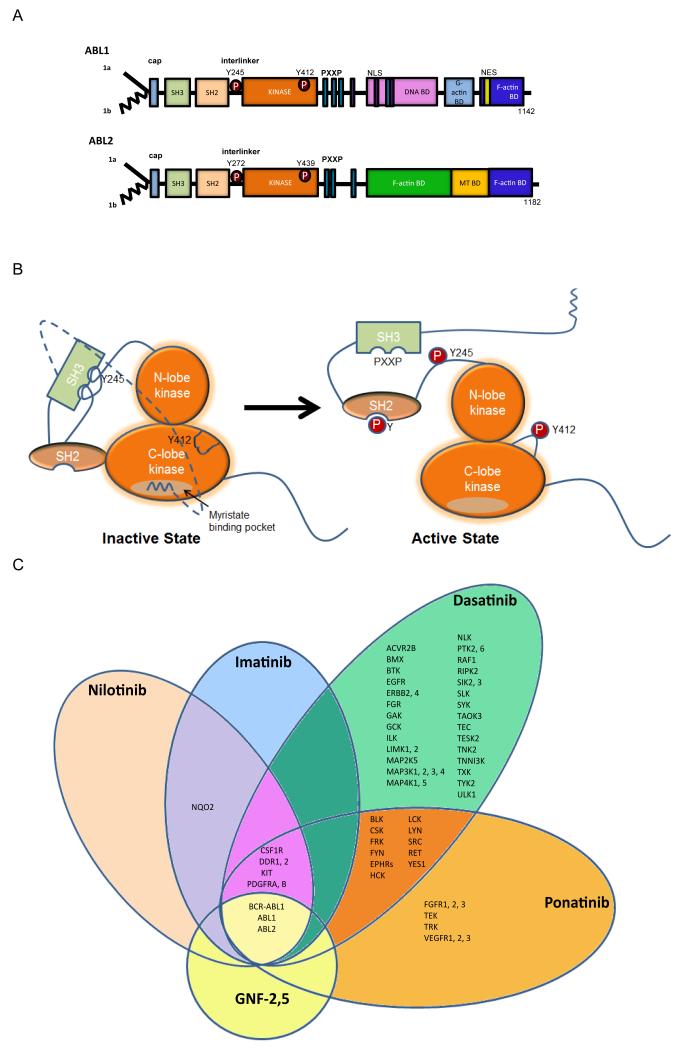
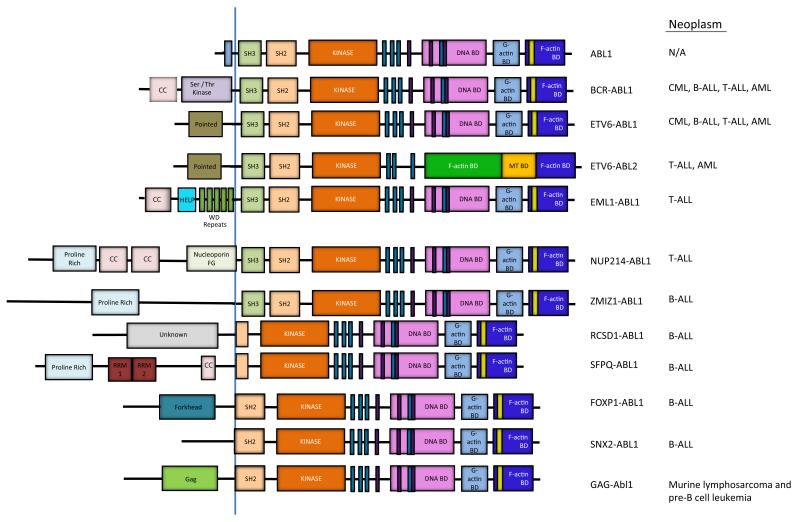
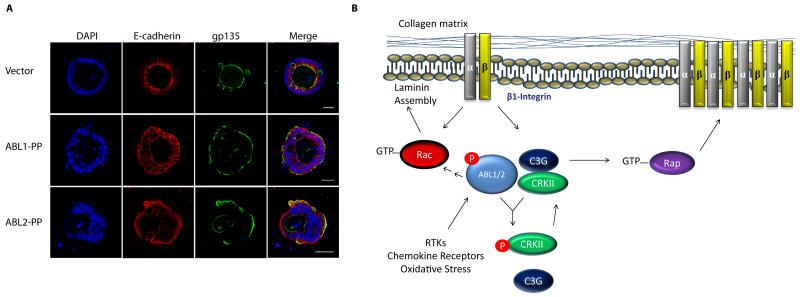
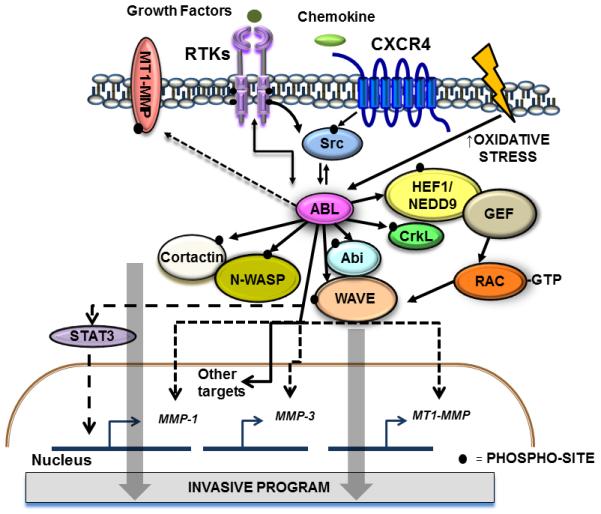
The Abelson (ABL) family of nonreceptor tyrosine kinases, ABL1 and ABL2, transduces diverse extracellular signals to protein networks that control proliferation, survival, migration and invasion. ABL1 was first identified as an oncogene required for the development of leukaemias initiated by retroviruses or chromosome translocations. The demonstration that small-molecule ABL kinase inhibitors could effectively treat chronic myeloid leukaemia opened the door to the era of targeted cancer therapies. Recent reports have uncovered roles for ABL kinases in solid tumours. Enhanced ABL expression and activation in some solid tumours, together with altered cell polarity, invasion or growth induced by activated ABL kinases, suggest that drugs targeting these kinases may be useful for treating selected solid tumours.
Figures
Similar articles
-
Multifunctional Abl kinases in health and disease.J Cell Sci. 2016 Jan 1;129(1):9-16. doi: 10.1242/jcs.175521. J Cell Sci. 2016. PMID: 26729027 Free PMC article. Review.
-
Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study.Lancet Haematol. 2021 Jan;8(1):e55-e66. doi: 10.1016/S2352-3026(20)30353-7. Epub 2020 Dec 22. Lancet Haematol. 2021. PMID: 33357483 Free PMC article. Clinical Trial.
-
The capable ABL: what is its biological function?Mol Cell Biol. 2014 Apr;34(7):1188-97. doi: 10.1128/MCB.01454-13. Epub 2014 Jan 13. Mol Cell Biol. 2014. PMID: 24421390 Free PMC article. Review.
-
Overcoming BCR::ABL1 dependent and independent survival mechanisms in chronic myeloid leukaemia using a multi-kinase targeting approach.Cell Commun Signal. 2023 Nov 29;21(1):342. doi: 10.1186/s12964-023-01363-2. Cell Commun Signal. 2023. PMID: 38031192 Free PMC article.
-
Targeting Abl kinases to regulate vascular leak during sepsis and acute respiratory distress syndrome.Arterioscler Thromb Vasc Biol. 2015 May;35(5):1071-9. doi: 10.1161/ATVBAHA.115.305085. Epub 2015 Mar 26. Arterioscler Thromb Vasc Biol. 2015. PMID: 25814671 Free PMC article. Review.
Cited by
-
Coordination of signalling networks and tumorigenic properties by ABL in glioblastoma cells.Oncotarget. 2016 Nov 15;7(46):74747-74767. doi: 10.18632/oncotarget.12546. Oncotarget. 2016. PMID: 27732969 Free PMC article.
-
Biomechanical regulation of breast cancer metastasis and progression.Sci Rep. 2021 May 10;11(1):9838. doi: 10.1038/s41598-021-89288-z. Sci Rep. 2021. PMID: 33972619 Free PMC article.
-
Identification of differentially expressed tumour-related genes regulated by UHRF1-driven DNA methylation.Sci Rep. 2024 Aug 7;14(1):18371. doi: 10.1038/s41598-024-69110-2. Sci Rep. 2024. PMID: 39112494 Free PMC article.
-
Cumulative mechanism of several major imatinib-resistant mutations in Abl kinase.Proc Natl Acad Sci U S A. 2020 Aug 11;117(32):19221-19227. doi: 10.1073/pnas.1919221117. Epub 2020 Jul 27. Proc Natl Acad Sci U S A. 2020. PMID: 32719139 Free PMC article.
-
α3β1 Integrin Suppresses Prostate Cancer Metastasis via Regulation of the Hippo Pathway.Cancer Res. 2016 Nov 15;76(22):6577-6587. doi: 10.1158/0008-5472.CAN-16-1483. Epub 2016 Sep 28. Cancer Res. 2016. PMID: 27680681 Free PMC article.
References
-
- Goff SP, Gilboa E, Witte ON, Baltimore D. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: studies with cloned viral DNA. Cell. 1980;22:777–85. - PubMed
-
-
Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233:212–4. References and 2 are historic manuscripts that describe the identification of normal and oncogenic forms of mouse and human ABL kinases.
-
-
- Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–91. - PubMed
-
- Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res. 2002;85:51–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
