

A comparative analysis of algorithms for somatic SNV detection in cancer
- PMID: 23842810
- PMCID: PMC3753564
- DOI: 10.1093/bioinformatics/btt375
A comparative analysis of algorithms for somatic SNV detection in cancer
Abstract
Motivation: With the advent of relatively affordable high-throughput technologies, DNA sequencing of cancers is now common practice in cancer research projects and will be increasingly used in clinical practice to inform diagnosis and treatment. Somatic (cancer-only) single nucleotide variants (SNVs) are the simplest class of mutation, yet their identification in DNA sequencing data is confounded by germline polymorphisms, tumour heterogeneity and sequencing and analysis errors. Four recently published algorithms for the detection of somatic SNV sites in matched cancer-normal sequencing datasets are VarScan, SomaticSniper, JointSNVMix and Strelka. In this analysis, we apply these four SNV calling algorithms to cancer-normal Illumina exome sequencing of a chronic myeloid leukaemia (CML) patient. The candidate SNV sites returned by each algorithm are filtered to remove likely false positives, then characterized and compared to investigate the strengths and weaknesses of each SNV calling algorithm.
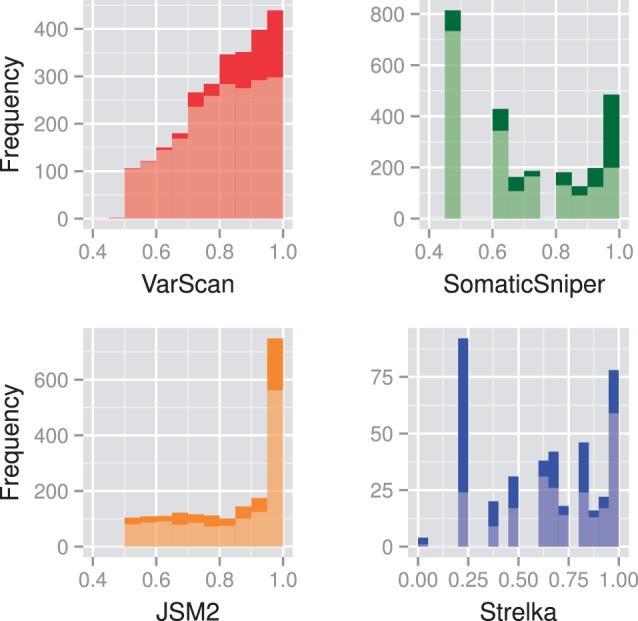
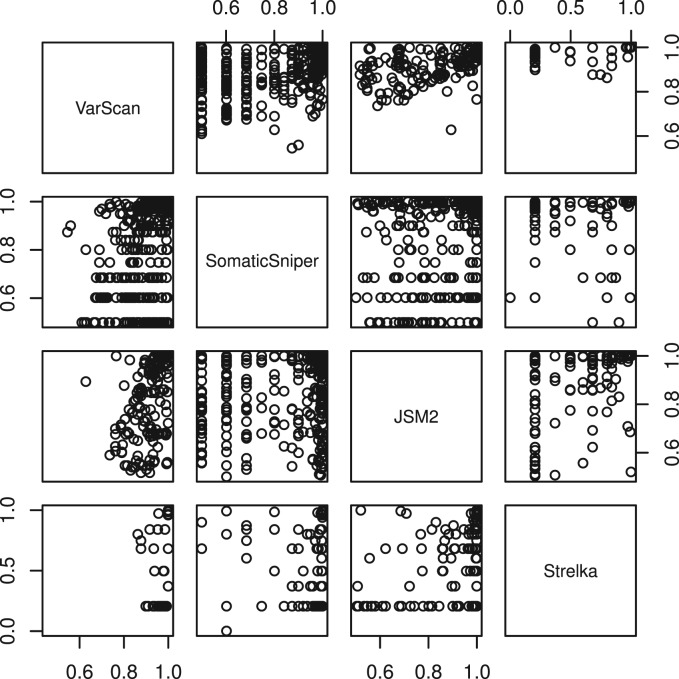
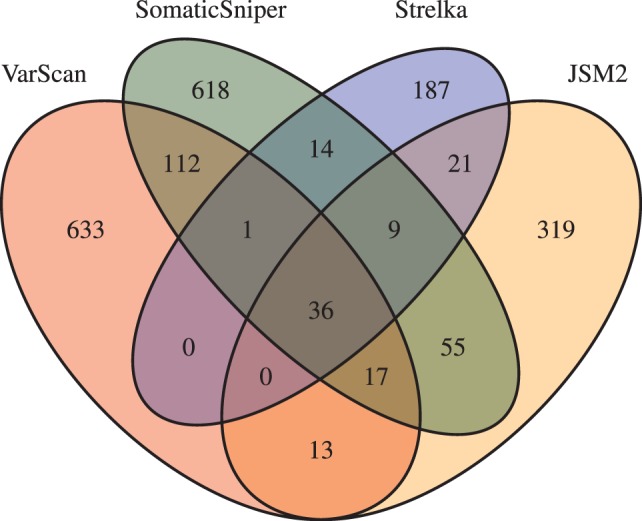
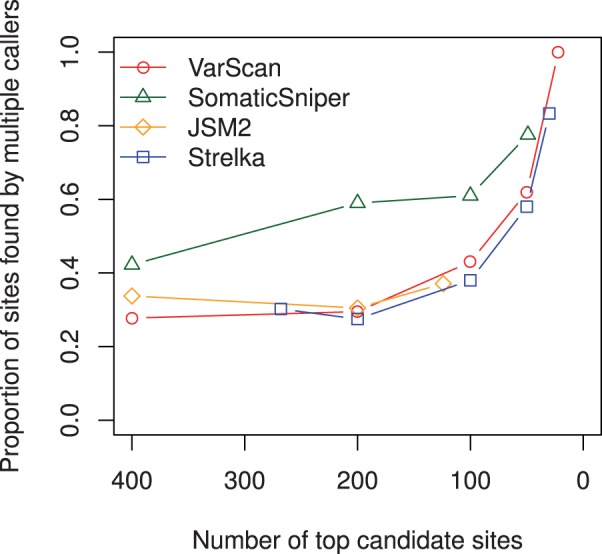
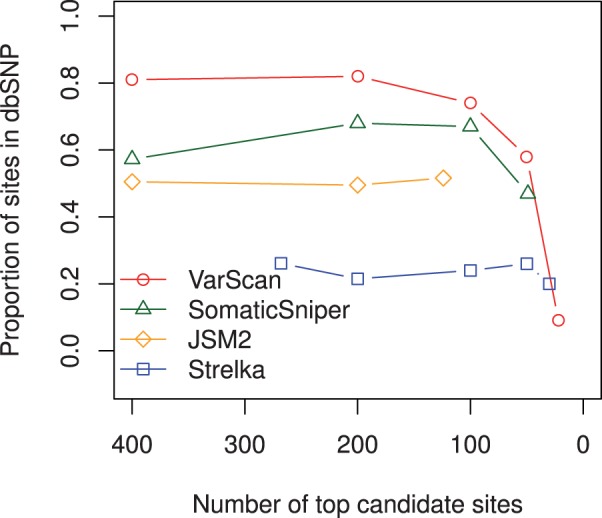
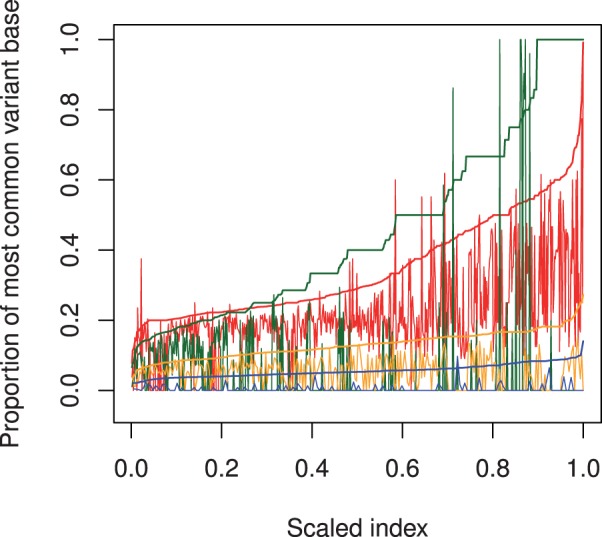
Results: Comparing the candidate SNV sets returned by VarScan, SomaticSniper, JointSNVMix2 and Strelka revealed substantial differences with respect to the number and character of sites returned; the somatic probability scores assigned to the same sites; their susceptibility to various sources of noise; and their sensitivities to low-allelic-fraction candidates.
Availability: Data accession number SRA081939, code at http://code.google.com/p/snv-caller-review/
Contact: david.adelson@adelaide.edu.au
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
