

Ground state destabilization by anionic nucleophiles contributes to the activity of phosphoryl transfer enzymes
- PMID: 23843744
- PMCID: PMC3699461
- DOI: 10.1371/journal.pbio.1001599
Ground state destabilization by anionic nucleophiles contributes to the activity of phosphoryl transfer enzymes
Abstract
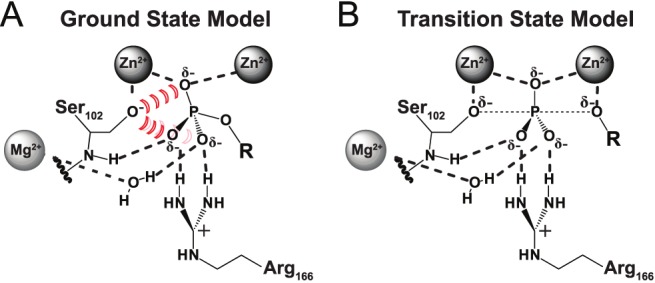
Enzymes stabilize transition states of reactions while limiting binding to ground states, as is generally required for any catalyst. Alkaline Phosphatase (AP) and other nonspecific phosphatases are some of Nature's most impressive catalysts, achieving preferential transition state over ground state stabilization of more than 10²²-fold while utilizing interactions with only the five atoms attached to the transferred phosphorus. We tested a model that AP achieves a portion of this preference by destabilizing ground state binding via charge repulsion between the anionic active site nucleophile, Ser102, and the negatively charged phosphate monoester substrate. Removal of the Ser102 alkoxide by mutation to glycine or alanine increases the observed Pi affinity by orders of magnitude at pH 8.0. To allow precise and quantitative comparisons, the ionic form of bound P(i) was determined from pH dependencies of the binding of Pi and tungstate, a P(i) analog lacking titratable protons over the pH range of 5-11, and from the ³¹P chemical shift of bound P(i). The results show that the Pi trianion binds with an exceptionally strong femtomolar affinity in the absence of Ser102, show that its binding is destabilized by ≥10⁸-fold by the Ser102 alkoxide, and provide direct evidence for ground state destabilization. Comparisons of X-ray crystal structures of AP with and without Ser102 reveal the same active site and P(i) binding geometry upon removal of Ser102, suggesting that the destabilization does not result from a major structural rearrangement upon mutation of Ser102. Analogous Pi binding measurements with a protein tyrosine phosphatase suggest the generality of this ground state destabilization mechanism. Our results have uncovered an important contribution of anionic nucleophiles to phosphoryl transfer catalysis via ground state electrostatic destabilization and an enormous capacity of the AP active site for specific and strong recognition of the phosphoryl group in the transition state.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
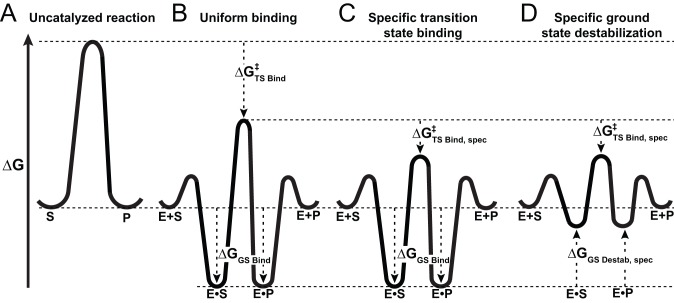
 ) so that the resulting reaction barrier is equal to the uncatalyzed reaction barrier under saturating conditions. This enzyme is not a catalyst as stabilization of the transition state without parallel stabilization of the ground state is required for catalysis. (C) Hypothetical enzyme that makes additional, specific transition state stabilizing interactions,
) so that the resulting reaction barrier is equal to the uncatalyzed reaction barrier under saturating conditions. This enzyme is not a catalyst as stabilization of the transition state without parallel stabilization of the ground state is required for catalysis. (C) Hypothetical enzyme that makes additional, specific transition state stabilizing interactions,  so that the reaction barrier between E.S and E.P is lower than that for the uncatalyzed barrier. This enzyme is a catalyst. (D) Hypothetical enzyme that makes specific ground state destabilizing interactions,
so that the reaction barrier between E.S and E.P is lower than that for the uncatalyzed barrier. This enzyme is a catalyst. (D) Hypothetical enzyme that makes specific ground state destabilizing interactions,  , to further enhance the catalytic properties of the enzyme relative to that in panel (C). This destabilization is discussed in the text.
, to further enhance the catalytic properties of the enzyme relative to that in panel (C). This destabilization is discussed in the text.
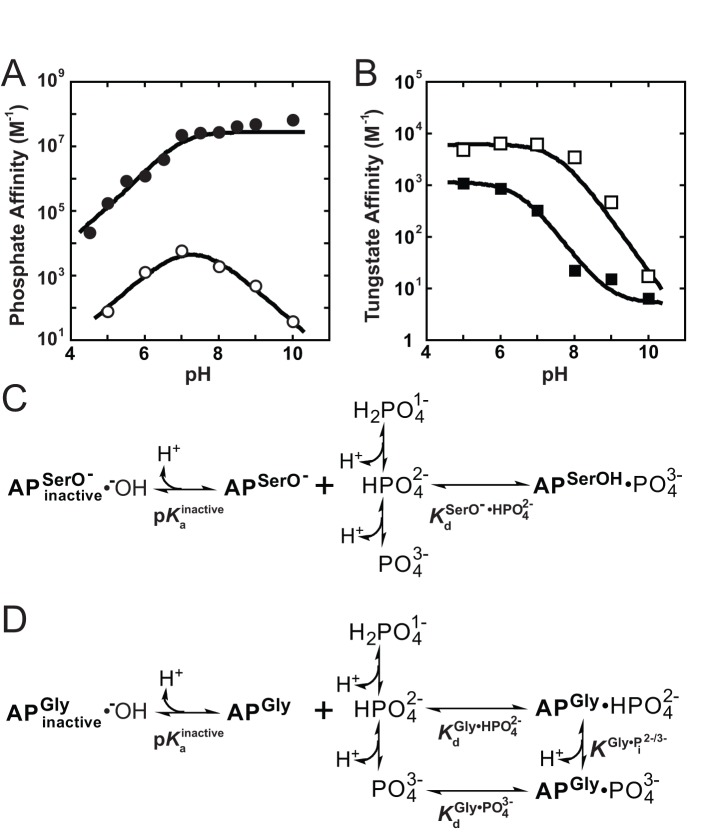
 = 7.6±0.1 and
= 7.6±0.1 and  = 110±20 µM. For S102G/R166S AP,
= 110±20 µM. For S102G/R166S AP,  was fixed at 6.5 based on the tungstate binding data in part (B) and
was fixed at 6.5 based on the tungstate binding data in part (B) and  was fixed at 10−6.1 M based on the 31P NMR data in Figure 6. Fits yielded
was fixed at 10−6.1 M based on the 31P NMR data in Figure 6. Fits yielded  = 93±8 nM, and
= 93±8 nM, and  = 210±20 fM. (B) The pH-dependent binding of tungstate to R166S (open squares) and S102G/R166S AP (closed squares). A weighted, nonlinear least-squares fit of
= 210±20 fM. (B) The pH-dependent binding of tungstate to R166S (open squares) and S102G/R166S AP (closed squares). A weighted, nonlinear least-squares fit of  derived from a two-state tungstate binding model (
derived from a two-state tungstate binding model ( ) yielded a fit of the R166S AP data with
) yielded a fit of the R166S AP data with  = 170±25 µM and
= 170±25 µM and  = 7.6±0.1. The corresponding fit to the observed tungstate affinity of S102G/R166S AP yielded a
= 7.6±0.1. The corresponding fit to the observed tungstate affinity of S102G/R166S AP yielded a  = 890±90 µM and
= 890±90 µM and  = 6.5±0.1. The tungstate affinity of S102G/R166S AP is weaker than R166S AP, indicating that Ser102 plays a favorable role in tungstate binding, possibly by allowing formation of octahedral tungstate as observed in other proteins that bind tungstate ,. At pH values ≥8 where the Pi affinity is strongest, the observed competition of 32Pi binding is likely influenced by competition from contaminating, unlabeled Pi in the tungstate stock rather than tungstate (see Materials and Methods). The dashed portion of the S102G/R166S AP tungstate fit line illustrates where the observed affinity can be accounted for by 0.5 ppm Pi contamination. Omitting the pH 9 and 10 points in the fit did not significantly change the fitted
= 6.5±0.1. The tungstate affinity of S102G/R166S AP is weaker than R166S AP, indicating that Ser102 plays a favorable role in tungstate binding, possibly by allowing formation of octahedral tungstate as observed in other proteins that bind tungstate ,. At pH values ≥8 where the Pi affinity is strongest, the observed competition of 32Pi binding is likely influenced by competition from contaminating, unlabeled Pi in the tungstate stock rather than tungstate (see Materials and Methods). The dashed portion of the S102G/R166S AP tungstate fit line illustrates where the observed affinity can be accounted for by 0.5 ppm Pi contamination. Omitting the pH 9 and 10 points in the fit did not significantly change the fitted  or
or  values. (C) Binding model used to fit the pH-dependent Pi affinity of R166S (and WT) AP. (D) Binding model used to fit the pH-dependent Pi affinity of S102G/R166S AP.
values. (C) Binding model used to fit the pH-dependent Pi affinity of R166S (and WT) AP. (D) Binding model used to fit the pH-dependent Pi affinity of S102G/R166S AP.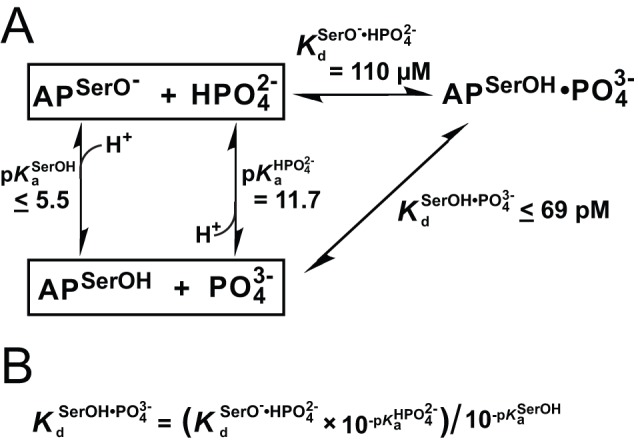
 for R166S AP is from a fit of the model in Figure 4C to the pH-dependent Pi binding affinity in Figure 4A. The Ser102 pK
a is an upper limit , and thus, the dissociation constant between protonated Ser102 and
for R166S AP is from a fit of the model in Figure 4C to the pH-dependent Pi binding affinity in Figure 4A. The Ser102 pK
a is an upper limit , and thus, the dissociation constant between protonated Ser102 and  (
( ) is also an upper limit (≤69 pM). The same cycle was used to establish an upper limit for the dissociation constant between WT AP with Ser102 protonated and
) is also an upper limit (≤69 pM). The same cycle was used to establish an upper limit for the dissociation constant between WT AP with Ser102 protonated and  ,
,  ≤290 fM, from the following values:
≤290 fM, from the following values:  ≤5.5,
≤5.5,  = 11.7, and
= 11.7, and  = 0.46 µM (Table 2). (B) Relationship derived from the thermodynamic cycle of part (A).
= 0.46 µM (Table 2). (B) Relationship derived from the thermodynamic cycle of part (A).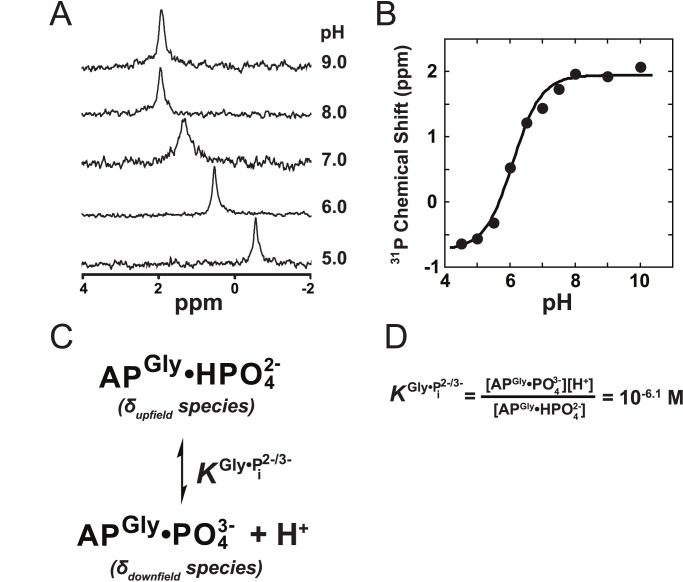
 of 10−6.1 M as defined in (D).
of 10−6.1 M as defined in (D).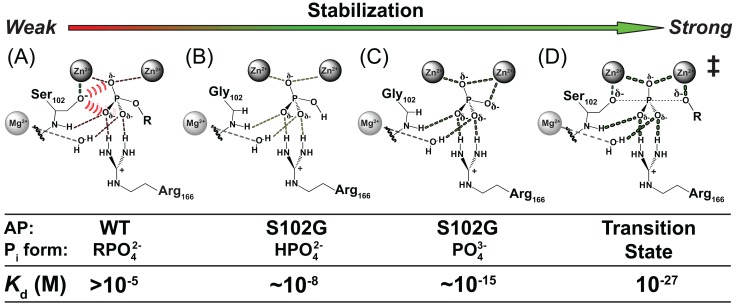
 was estimated to be ∼10 nM (Table 2). (C) Trianion binding (
was estimated to be ∼10 nM (Table 2). (C) Trianion binding ( ; Table 2) was estimated to be ∼1 fM and is 107-fold stronger than dianion binding. (D) The AP rate enhancement is 1027-fold (for methyl phosphate dianion hydrolysis: k
cat/K
M = 1.2×106 M−1 s−1
and k
uncat∼4×10−22 M−1 s−1
[75]), corresponding to a theoretical dissociation constant for transition state binding of 10−12 fM (derived in Figure S1 of ref. [16]). This theoretical affinity reflects binding of the enzyme to the transition state while accompanied by replacement of water by the active site Ser102 nucleophile. The energetics of these two processes cannot be separated and the formal dissociation constant reflects contributions from both binding interactions and positioning of the Ser102 nucleophile and substrate.
; Table 2) was estimated to be ∼1 fM and is 107-fold stronger than dianion binding. (D) The AP rate enhancement is 1027-fold (for methyl phosphate dianion hydrolysis: k
cat/K
M = 1.2×106 M−1 s−1
and k
uncat∼4×10−22 M−1 s−1
[75]), corresponding to a theoretical dissociation constant for transition state binding of 10−12 fM (derived in Figure S1 of ref. [16]). This theoretical affinity reflects binding of the enzyme to the transition state while accompanied by replacement of water by the active site Ser102 nucleophile. The energetics of these two processes cannot be separated and the formal dissociation constant reflects contributions from both binding interactions and positioning of the Ser102 nucleophile and substrate.Comment in
-
Don't get too comfortable: destabilizing the ground state to speed a reaction.PLoS Biol. 2013 Jul;11(7):e1001600. doi: 10.1371/journal.pbio.1001600. Epub 2013 Jul 2. PLoS Biol. 2013. PMID: 23843745 Free PMC article. No abstract available.
References
-
- Edwards DR, Lohman DC, Wolfenden R (2011) Catalytic proficiency: the extreme case of S-O cleaving sulfatases. J Am Chem Soc 134: 525–531. - PubMed
-
- Wolfenden R, Snider MJ (2001) The depth of chemical time and the power of enzymes as catalysts. Acc Chem Res 34: 938–945. - PubMed
-
- Bar-Even A, Noor E, Savir Y, Liebermeister W, Davidi D, et al. (2011) The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50: 4402–4410. - PubMed
-
- Jencks WP (1975) Binding energy, specificity, and enzymic catalysis: Circe effect. Adv Enzymol Relat Areas Mol Biol 43: 219–410. - PubMed
-
- Jencks WP (1987) Economics of enzyme catalysis. Cold Spring Harb Symp Quant Biol 52: 65–73. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
