

Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions
- PMID: 23844038
- PMCID: PMC3701070
- DOI: 10.1371/journal.pone.0067583
Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions
Abstract
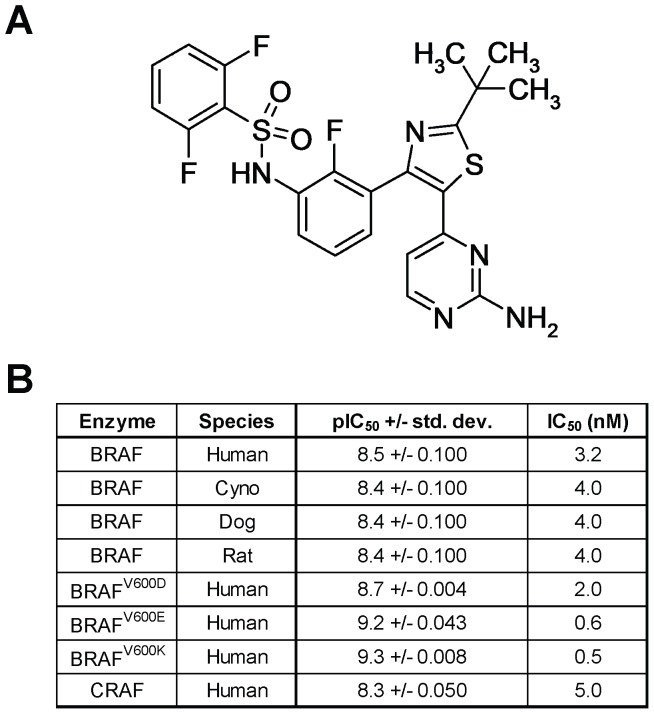
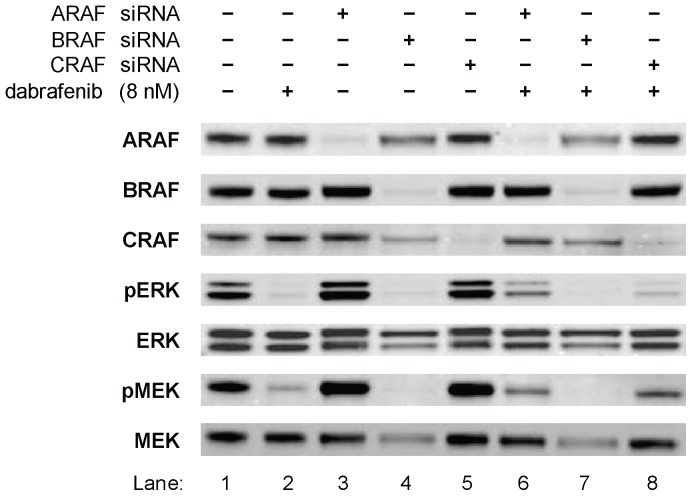
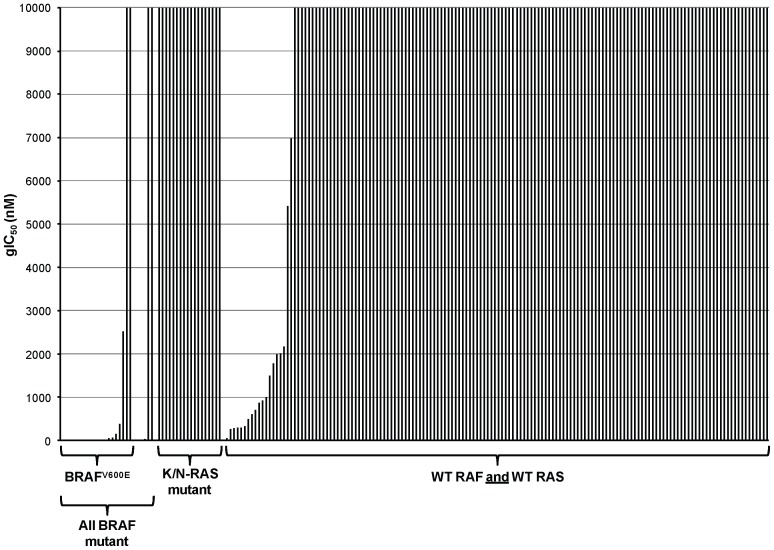
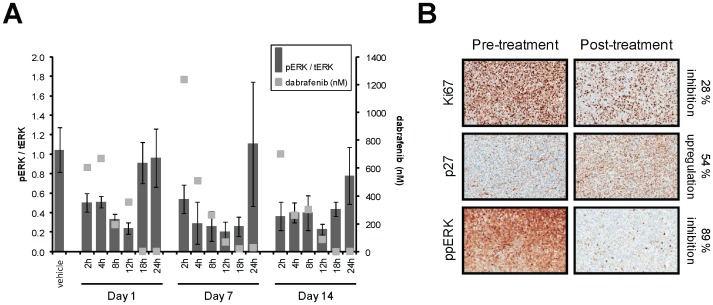
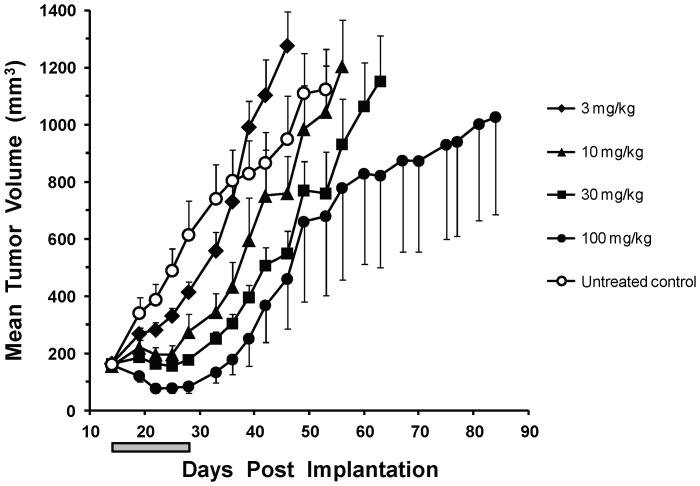
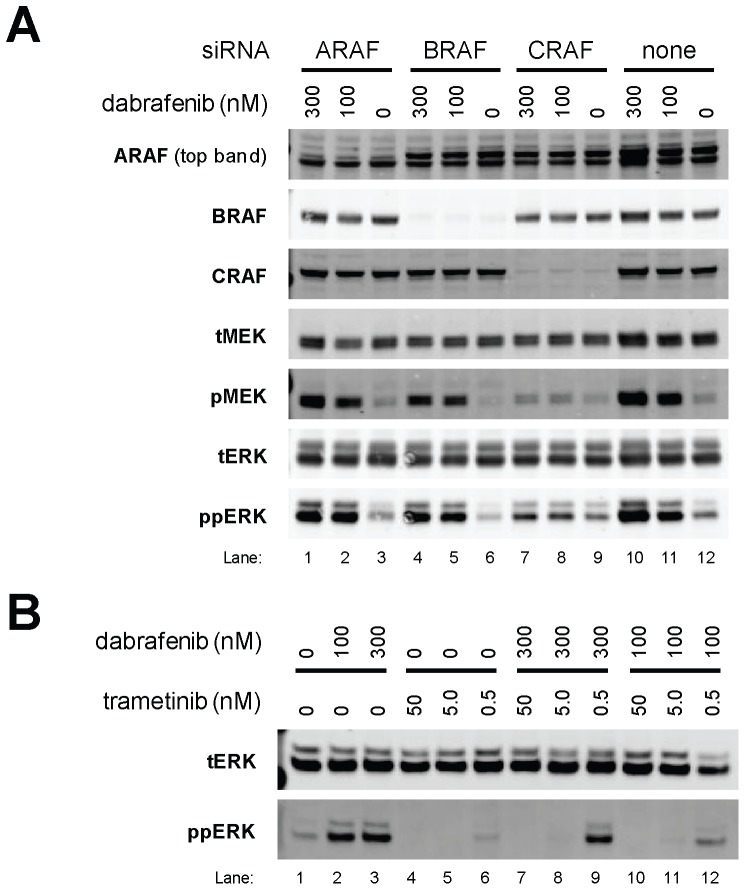
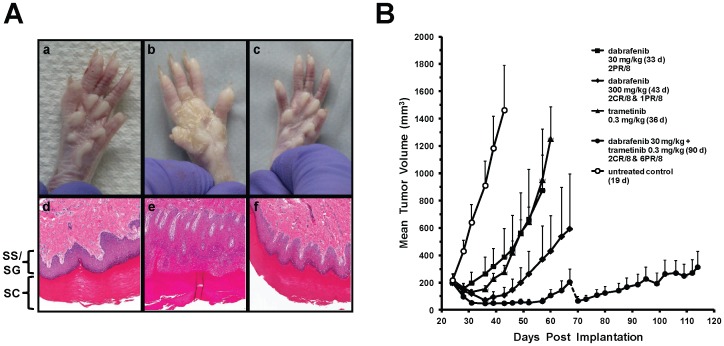
Mitogen-Activated Protein Kinase (MAPK) pathway activation has been implicated in many types of human cancer. BRAF mutations that constitutively activate MAPK signalling and bypass the need for upstream stimuli occur with high prevalence in melanoma, colorectal carcinoma, ovarian cancer, papillary thyroid carcinoma, and cholangiocarcinoma. In this report we characterize the novel, potent, and selective BRAF inhibitor, dabrafenib (GSK2118436). Cellular inhibition of BRAF(V600E) kinase activity by dabrafenib resulted in decreased MEK and ERK phosphorylation and inhibition of cell proliferation through an initial G1 cell cycle arrest, followed by cell death. In a BRAF(V600E)-containing xenograft model of human melanoma, orally administered dabrafenib inhibited ERK activation, downregulated Ki67, and upregulated p27, leading to tumor growth inhibition. However, as reported for other BRAF inhibitors, dabrafenib also induced MAPK pathway activation in wild-type BRAF cells through CRAF (RAF1) signalling, potentially explaining the squamous cell carcinomas and keratoacanthomas arising in patients treated with BRAF inhibitors. In addressing this issue, we showed that concomitant administration of BRAF and MEK inhibitors abrogated paradoxical BRAF inhibitor-induced MAPK signalling in cells, reduced the occurrence of skin lesions in rats, and enhanced the inhibition of human tumor xenograft growth in mouse models. Taken together, our findings offer preclinical proof of concept for dabrafenib as a specific and highly efficacious BRAF inhibitor and provide evidence for its potential clinical benefits when used in combination with a MEK inhibitor.
Conflict of interest statement
Figures
References
-
- Yoon S, Seger R (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24: 21–44. - PubMed
-
- Dhillon AS, Hagan S, Rath O, Kolch W (2007) MAP kinase signalling pathways in cancer. Oncogene 26: 3279–3290. - PubMed
-
- Montagut C, Settleman J (2009) Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett 283: 125–134. - PubMed
-
- Young A, Lyons J, Miller AL, Phan VT, Alarcon IR, et al. (2009) Ras signaling and therapies. Adv Cancer Res 102: 1–17. - PubMed
-
- Eggermont AM, Robert C (2011) New drugs in melanoma: it's a whole new world. Eur J Cancer 47: 2150–2157. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
