

The non-canonical Wnt/PKC pathway regulates mitochondrial dynamics through degradation of the arm-like domain-containing protein Alex3
- PMID: 23844091
- PMCID: PMC3699457
- DOI: 10.1371/journal.pone.0067773
The non-canonical Wnt/PKC pathway regulates mitochondrial dynamics through degradation of the arm-like domain-containing protein Alex3
Abstract
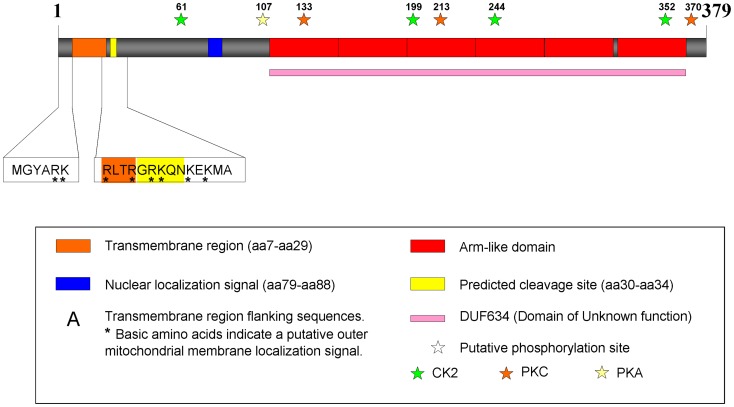
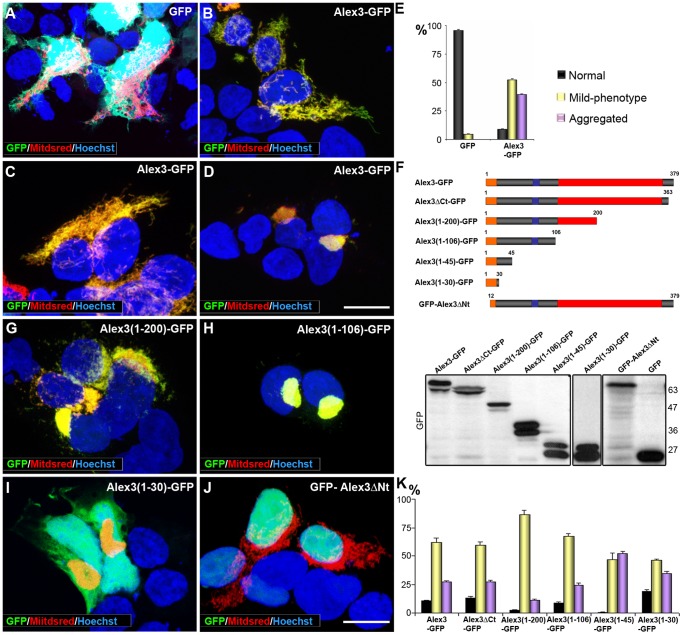
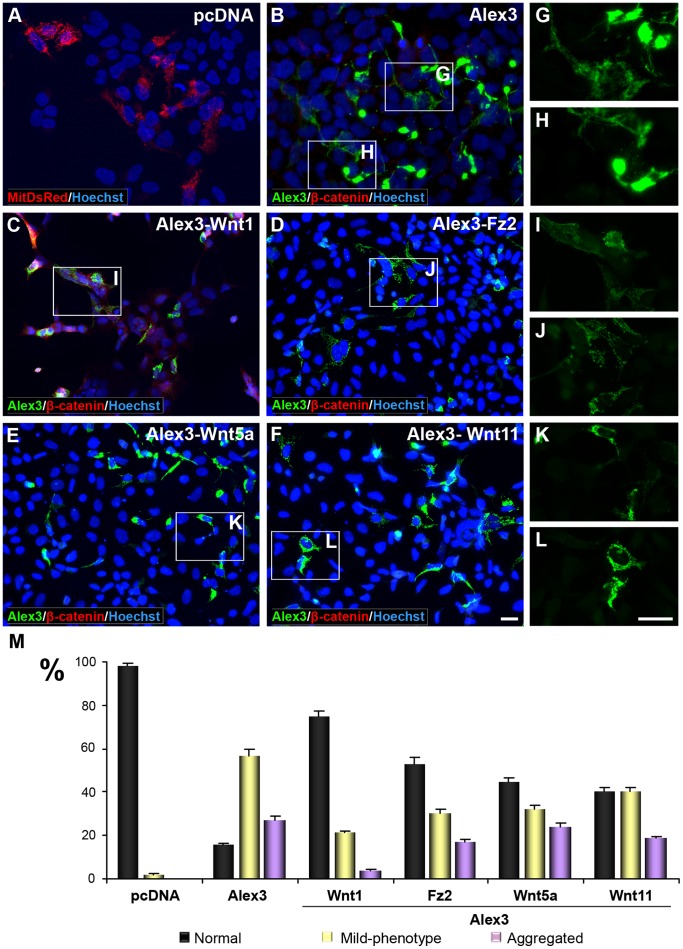
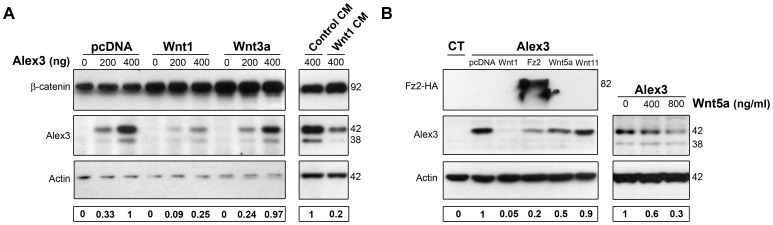
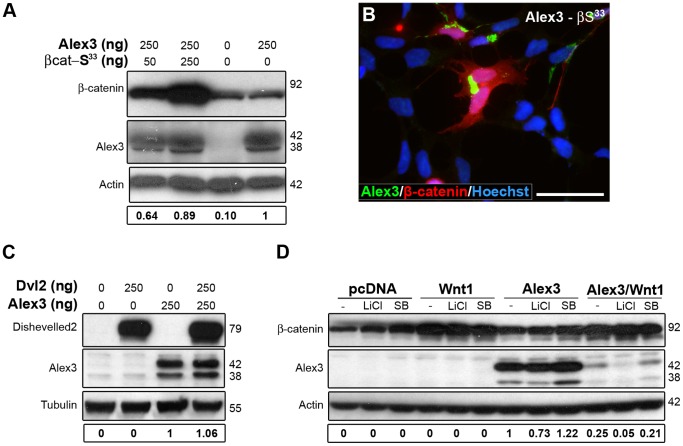
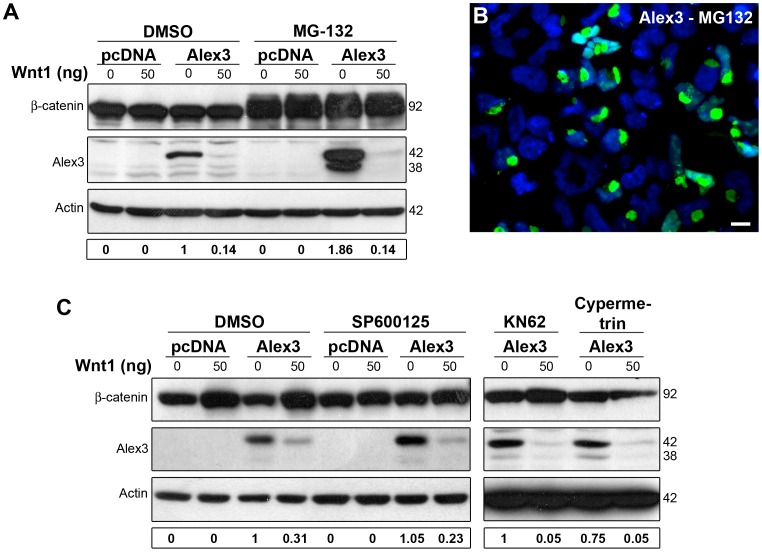
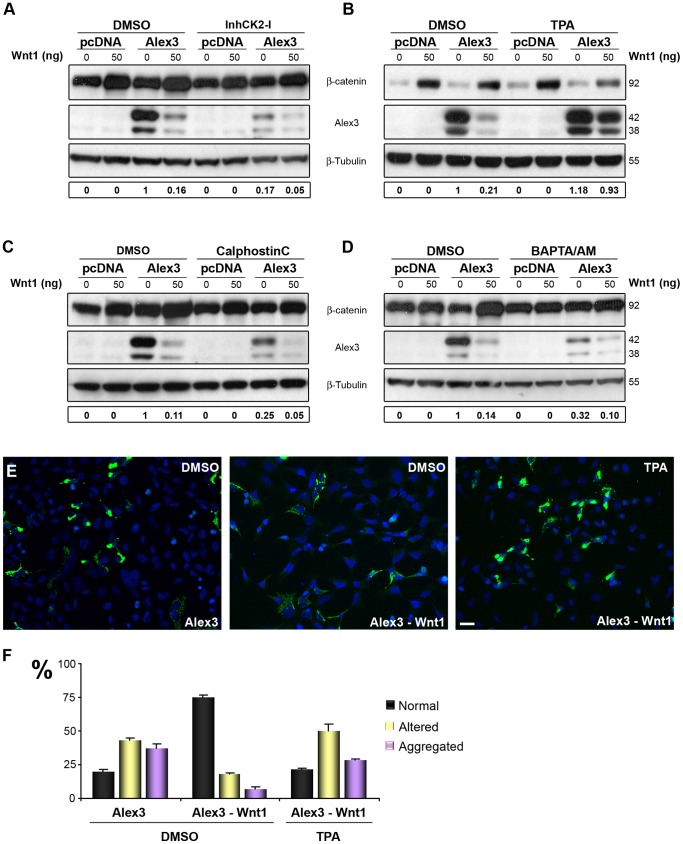
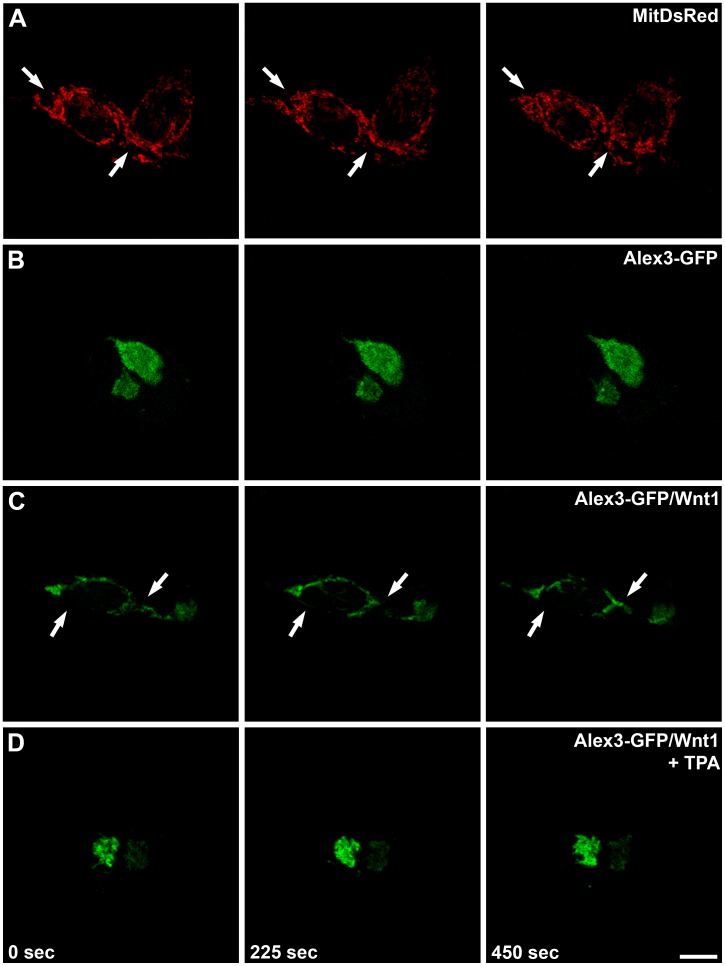
The regulation of mitochondrial dynamics is vital in complex cell types, such as neurons, that transport and localize mitochondria in high energy-demanding cell domains. The Armcx3 gene encodes a mitochondrial-targeted protein (Alex3) that contains several arm-like domains. In a previous study we showed that Alex3 protein regulates mitochondrial aggregation and trafficking. Here we studied the contribution of Wnt proteins to the mitochondrial aggregation and dynamics regulated by Alex3. Overexpression of Alex3 in HEK293 cells caused a marked aggregation of mitochondria, which was attenuated by treatment with several Wnts. We also found that this decrease was caused by Alex3 degradation induced by Wnts. While the Wnt canonical pathway did not alter the pattern of mitochondrial aggregation induced by Alex3, we observed that the Wnt/PKC non-canonical pathway regulated both mitochondrial aggregation and Alex3 protein levels, thereby rendering a mitochondrial phenotype and distribution similar to control patterns. Our data suggest that the Wnt pathway regulates mitochondrial distribution and dynamics through Alex3 protein degradation.
Conflict of interest statement
Figures
References
-
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315–360. - PubMed
-
- Han XJ, Tomizawa K, Fujimura A, Ohmori I, Nishiki T, et al. (2010) Regulation of mitochondrial dynamics and neurodegenerative diseases. Acta Med Okayama 65: 1–10. - PubMed
-
- Lopez-Domenech G, Serrat R, Mirra S, D'Aniello S, Somorjai I, et al. (2012) The Eutherian Armcx genes regulate mitochondrial trafficking in neurons and interact with Miro and Trak2. Nat Commun 3: 814. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
