

jMOSAiCS: joint analysis of multiple ChIP-seq datasets
- PMID: 23844871
- PMCID: PMC4053760
- DOI: 10.1186/gb-2013-14-4-r38
jMOSAiCS: joint analysis of multiple ChIP-seq datasets
Abstract
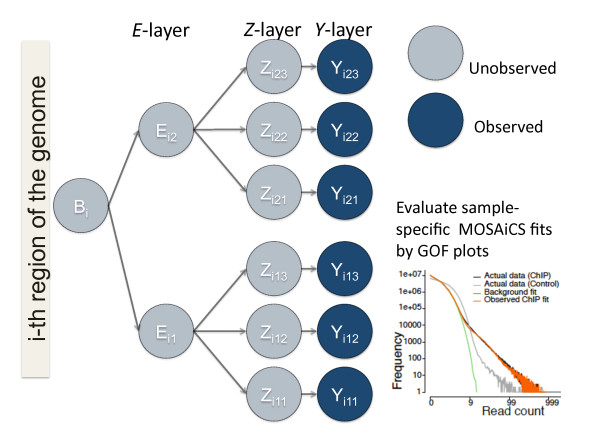
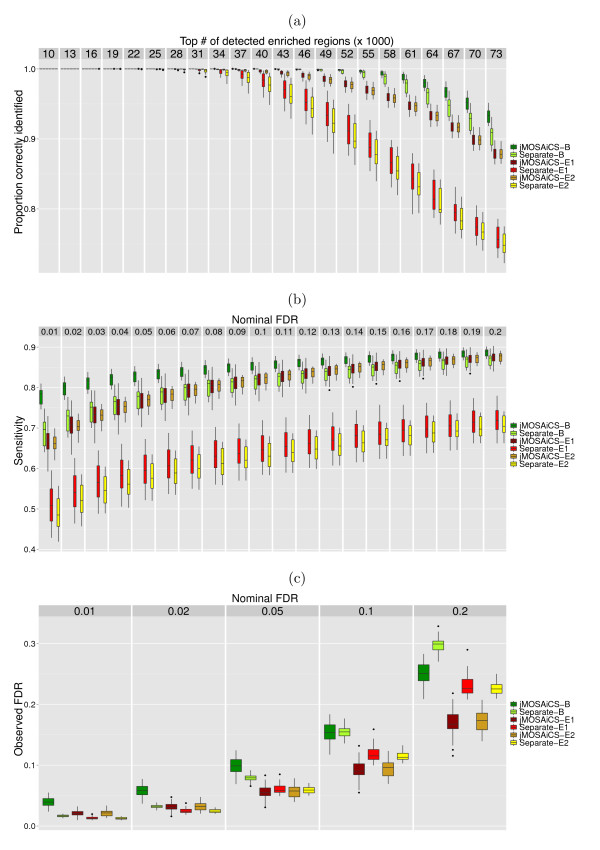
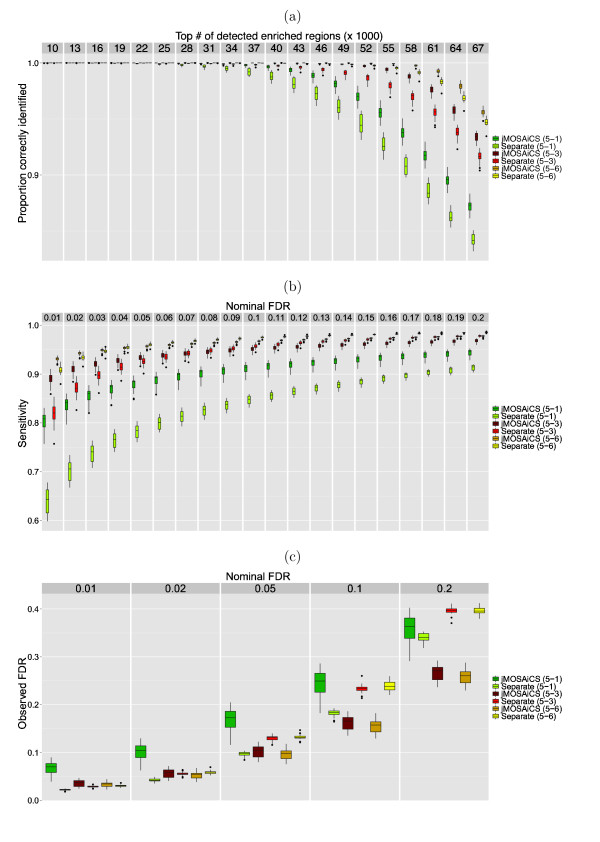
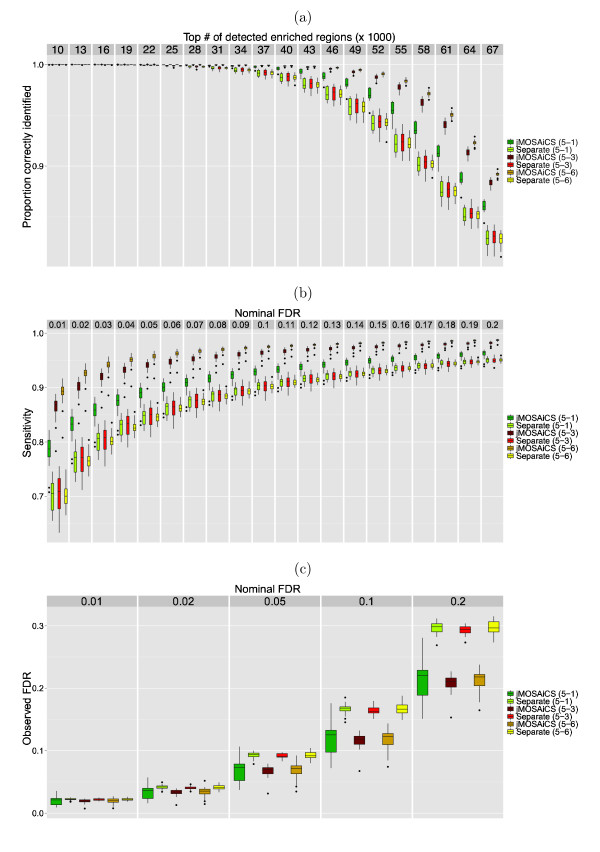
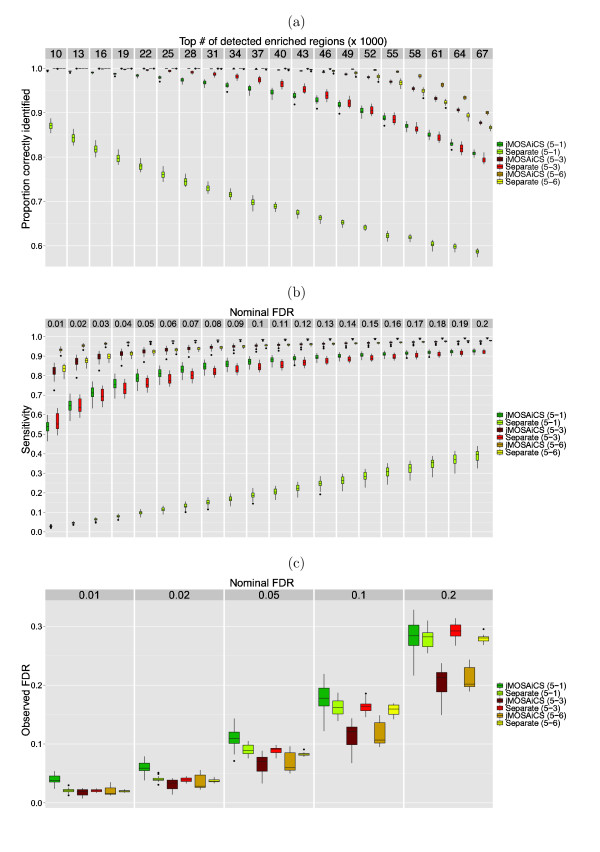
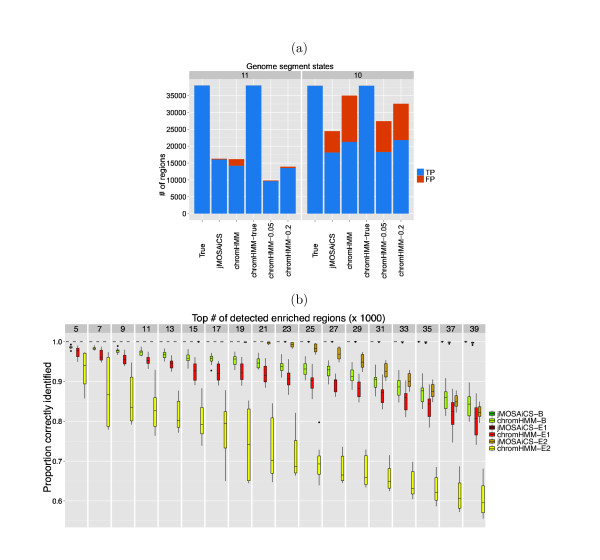
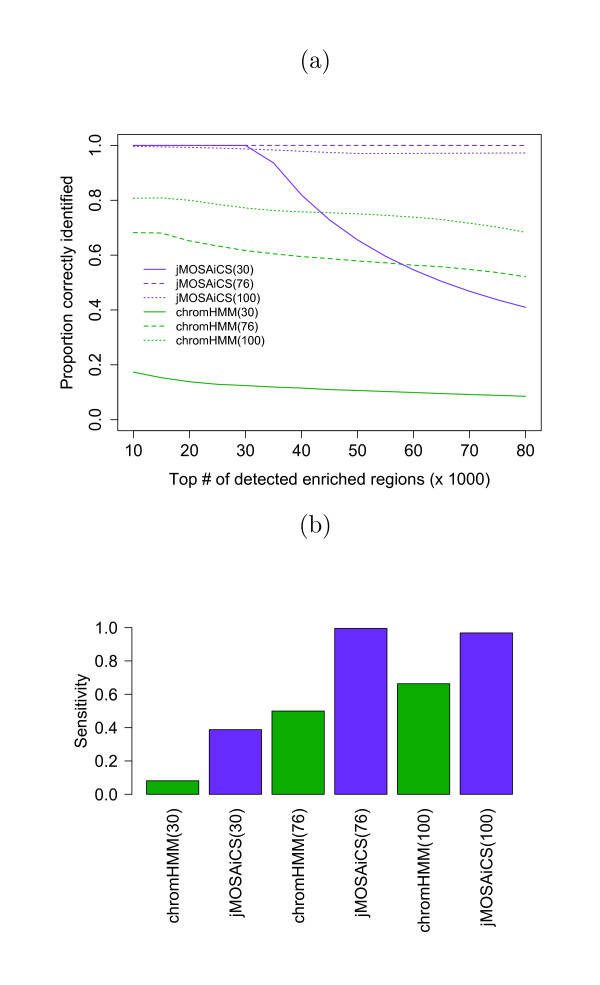
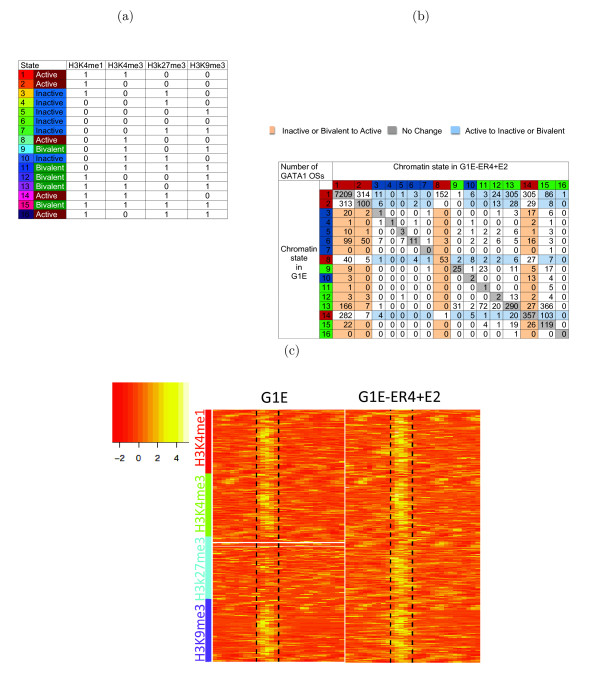
The ChIP-seq technique enables genome-wide mapping of in vivo protein-DNA interactions and chromatin states. Current analytical approaches for ChIP-seq analysis are largely geared towards single-sample investigations, and have limited applicability in comparative settings that aim to identify combinatorial patterns of enrichment across multiple datasets. We describe a novel probabilistic method, jMOSAiCS, for jointly analyzing multiple ChIP-seq datasets. We demonstrate its usefulness with a wide range of data-driven computational experiments and with a case study of histone modifications on GATA1-occupied segments during erythroid differentiation. jMOSAiCS is open source software and can be downloaded from Bioconductor 1.
Figures
References
-
- Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini A, Sawitzki G, Smith C, Smyth G, Tierney L, Yang J, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology. 2004;14:R80+. http://www.bioconductor.org/packages/devel/bioc/html/jmosaics.html - PMC - PubMed
-
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, Hong MY, Karczewski KJ, Huber W, Weissman SM, Gerstein MB, Korbel JO, Snyder M. Variation in transcription factor binding among humans. Science. 2010;14:232–235. doi: 10.1126/science.1183621. - DOI - PMC - PubMed
-
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, Alves P, Chateigner A, Perry M, Morris M, Auerbach RK, Feng X, Leng J, Vielle A, Niu W, Rhrissorrakrai K, Agarwal A, Alexander RP, Barber G, Brdlik CM, Brennan J, Brouillet JJ, Carr A, Cheung MS, Clawson H, Contrino S. et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;14:1775–1787. doi: 10.1126/science.1196914. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
