

A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention
- PMID: 23845441
- PMCID: PMC3706745
- DOI: 10.1016/j.ccr.2013.05.014
A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention
Abstract
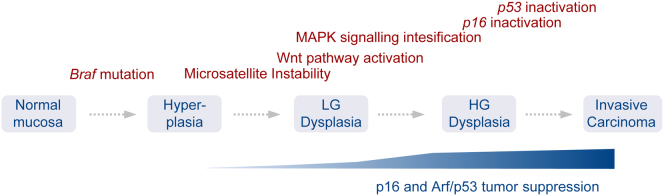
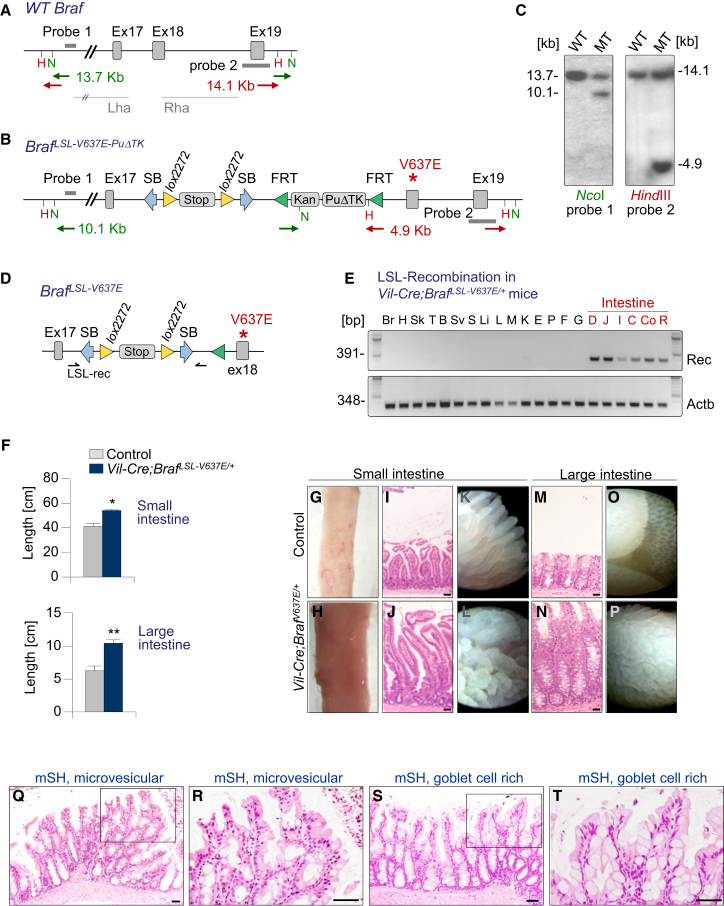
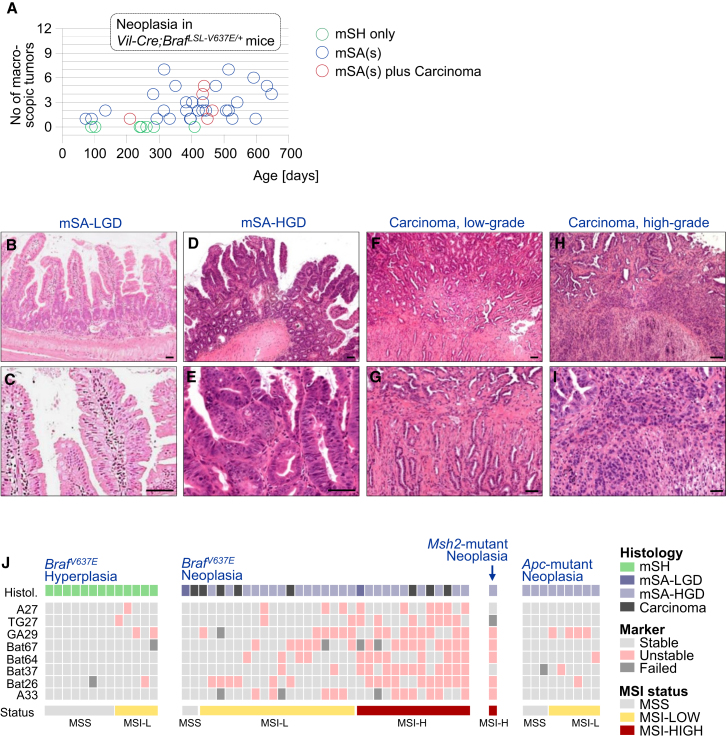
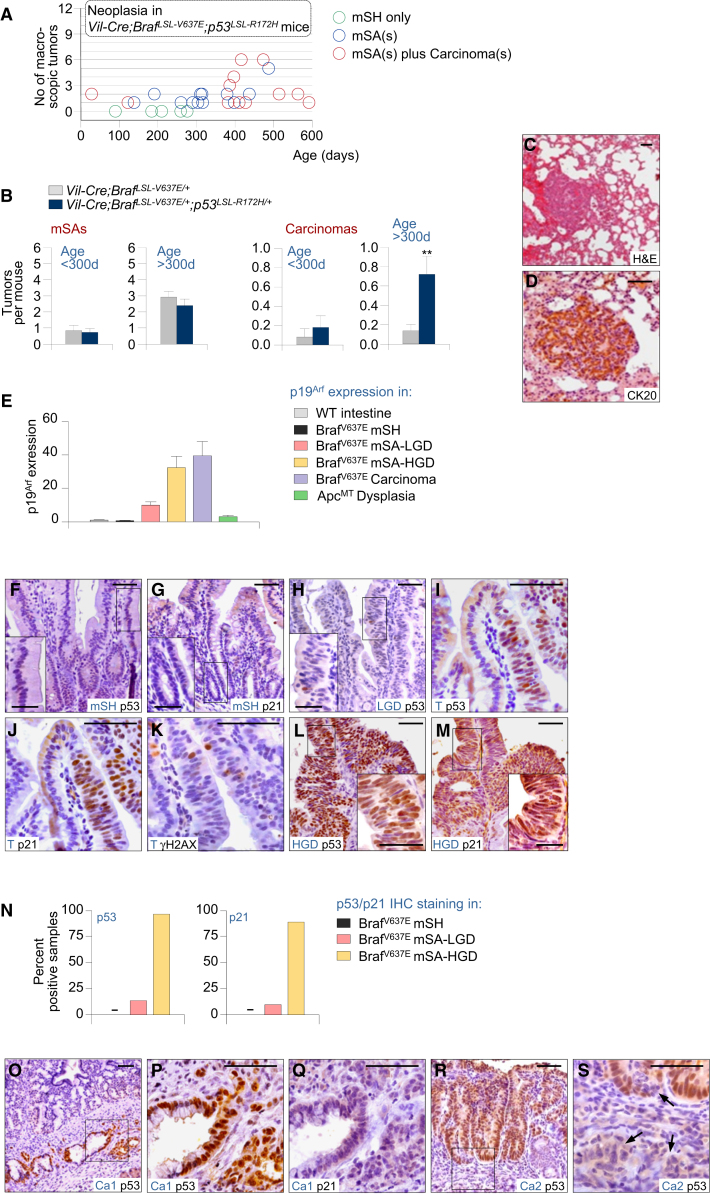
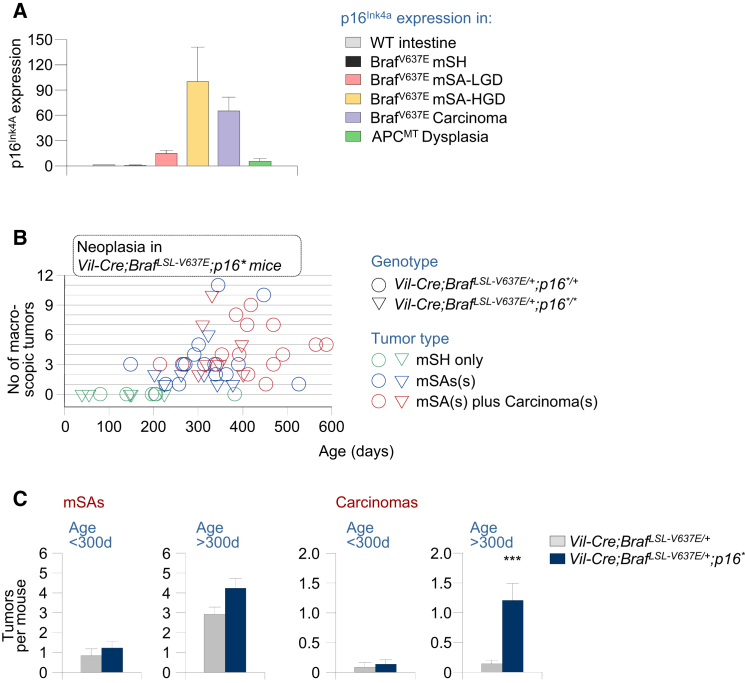
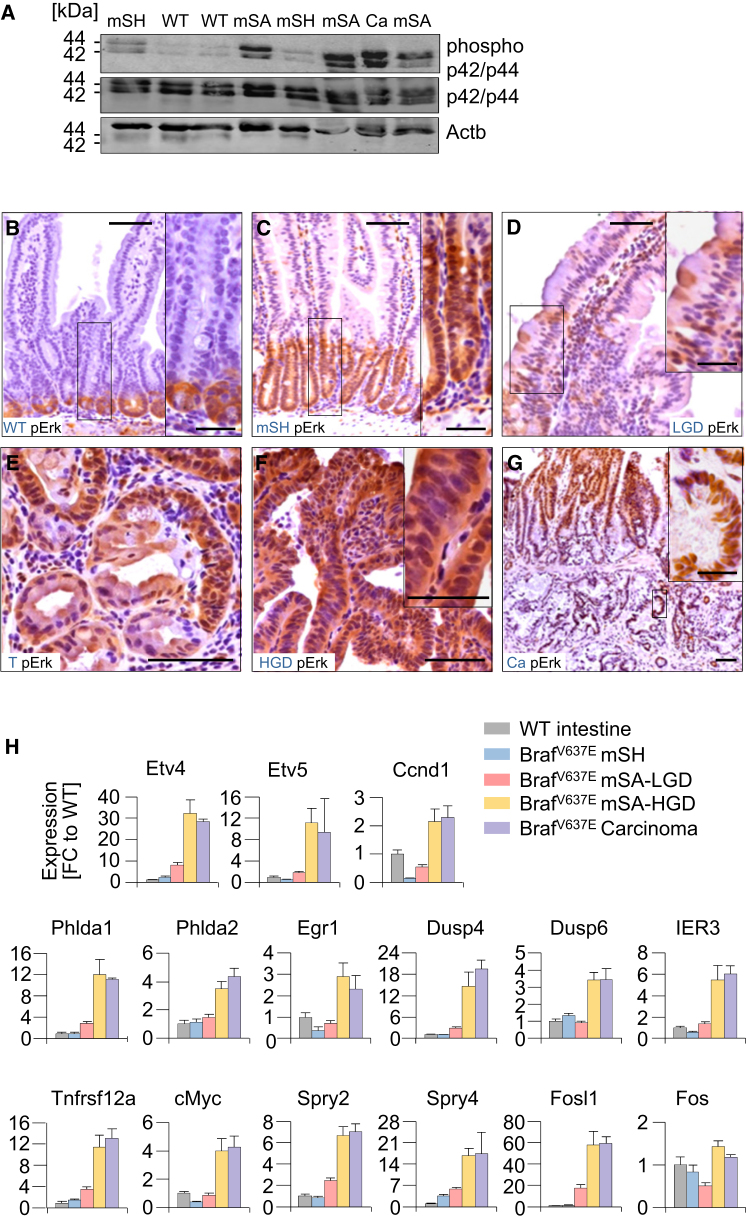
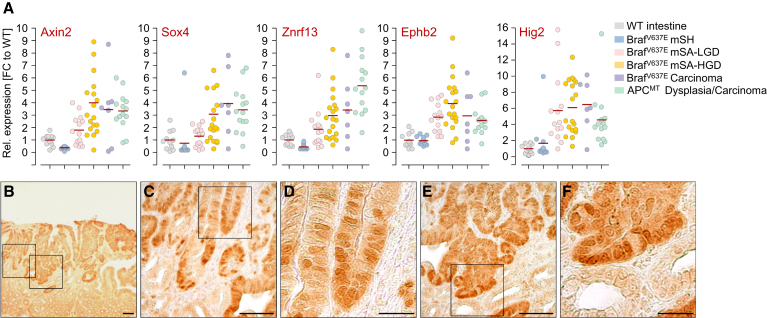
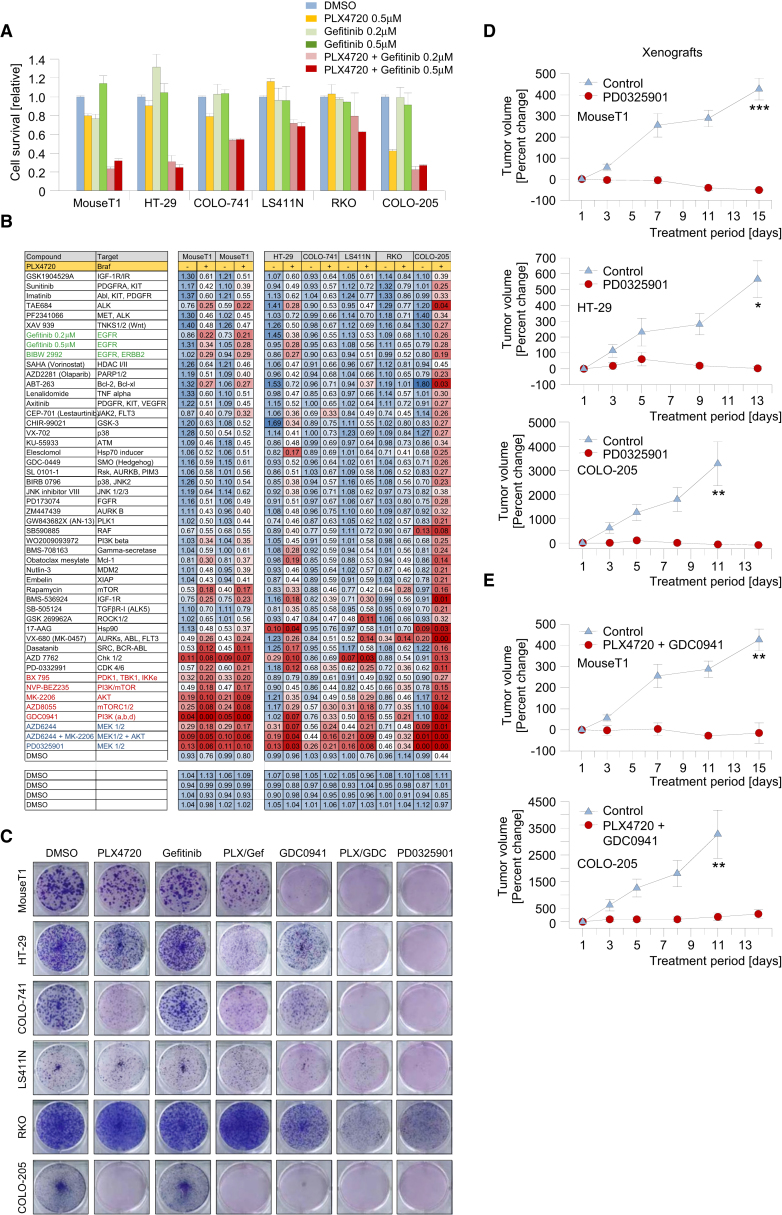
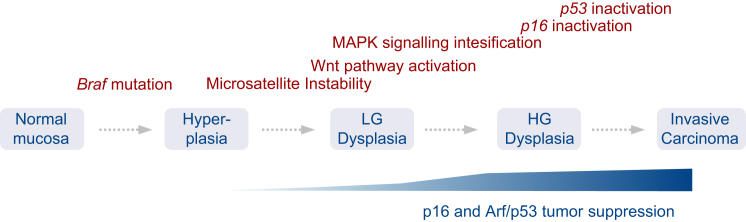
We show that BRAF(V600E) initiates an alternative pathway to colorectal cancer (CRC), which progresses through a hyperplasia/adenoma/carcinoma sequence. This pathway underlies significant subsets of CRCs with distinctive pathomorphologic/genetic/epidemiologic/clinical characteristics. Genetic and functional analyses in mice revealed a series of stage-specific molecular alterations driving different phases of tumor evolution and uncovered mechanisms underlying this stage specificity. We further demonstrate dose-dependent effects of oncogenic signaling, with physiologic Braf(V600E) expression being sufficient for hyperplasia induction, but later stage intensified Mapk-signaling driving both tumor progression and activation of intrinsic tumor suppression. Such phenomena explain, for example, the inability of p53 to restrain tumor initiation as well as its importance in invasiveness control, and the late stage specificity of its somatic mutation. Finally, systematic drug screening revealed sensitivity of this CRC subtype to targeted therapeutics, including Mek or combinatorial PI3K/Braf inhibition.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Bennecke M., Kriegl L., Bajbouj M., Retzlaff K., Robine S., Jung A., Arkan M.C., Kirchner T., Greten F.R. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–146. - PubMed
-
- Bettington M., Walker N., Clouston A., Brown I., Leggett B., Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–386. - PubMed
-
- Bond C.E., Umapathy A., Ramsnes I., Greco S.A., Zhao Z.Z., Mallitt K.A., Buttenshaw R.L., Montgomery G.W., Leggett B.A., Whitehall V.L. P53 mutation is common in microsatellite stable, BRAF mutant colorectal cancers. Int. J. Cancer. 2011;130:1567–1576. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
