

Suppression of inflammatory cascade is implicated in methyl amooranin-mediated inhibition of experimental mammary carcinogenesis
- PMID: 23846978
- PMCID: PMC3888835
- DOI: 10.1002/mc.22067
Suppression of inflammatory cascade is implicated in methyl amooranin-mediated inhibition of experimental mammary carcinogenesis
Abstract
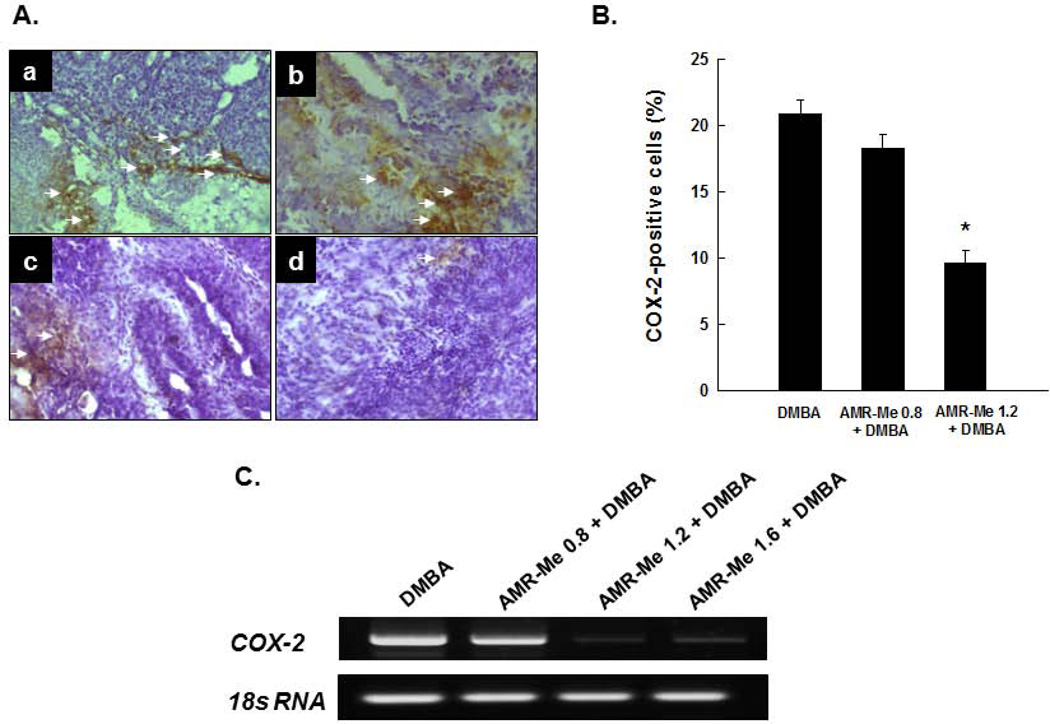
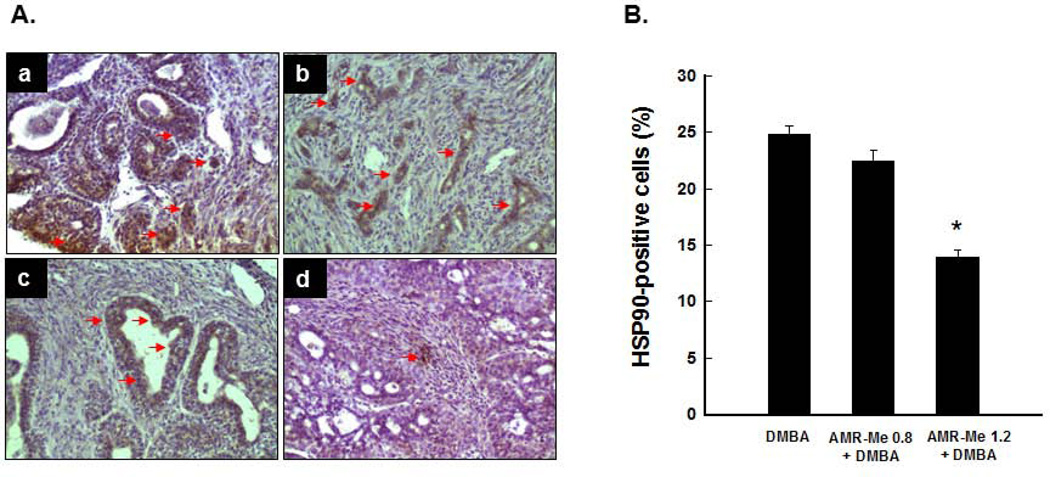
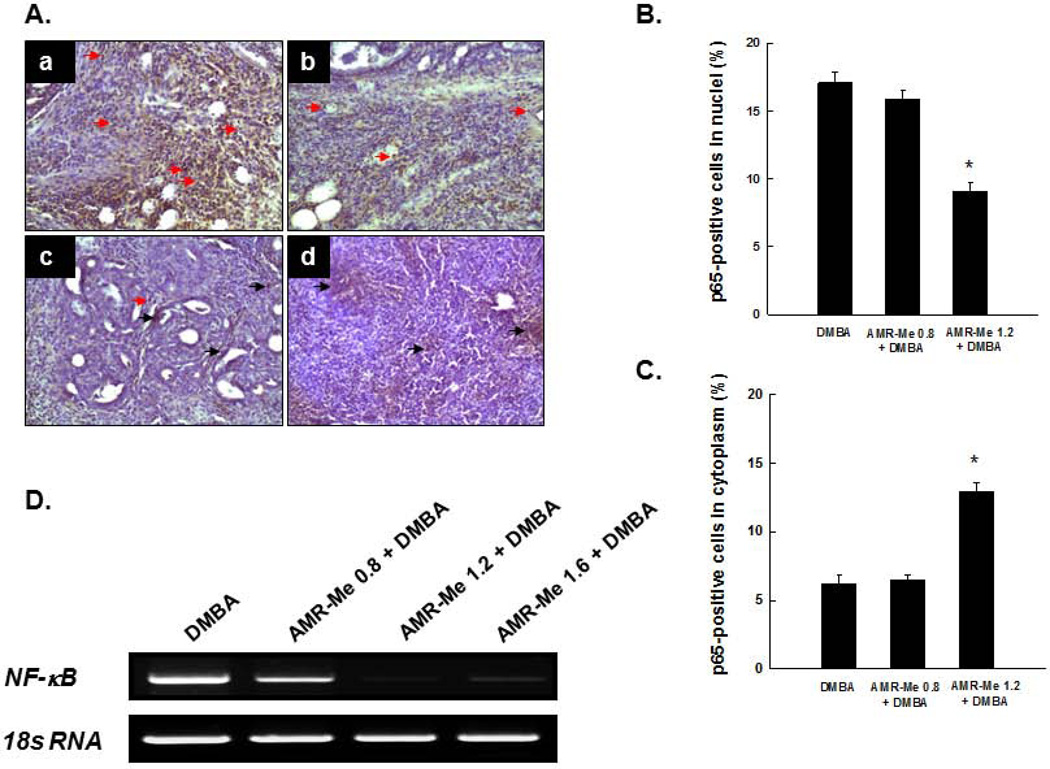
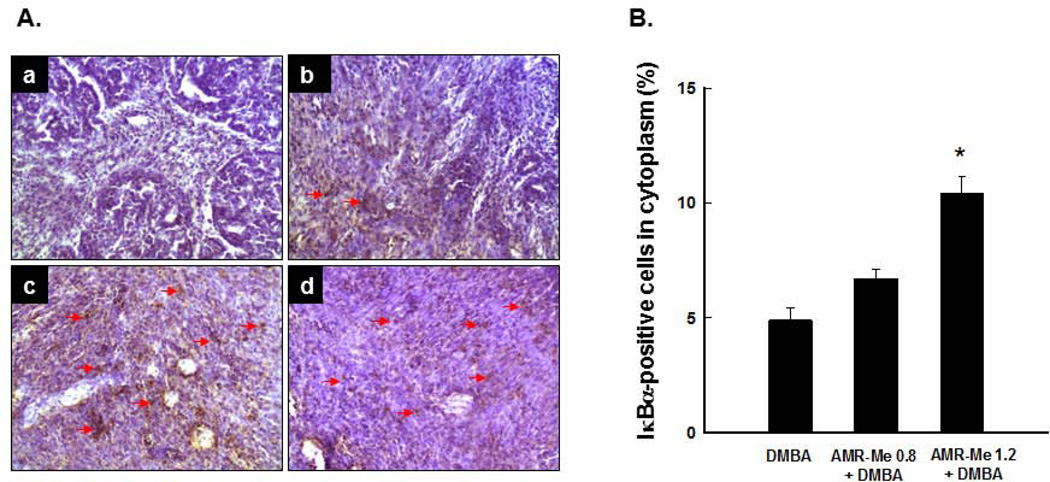
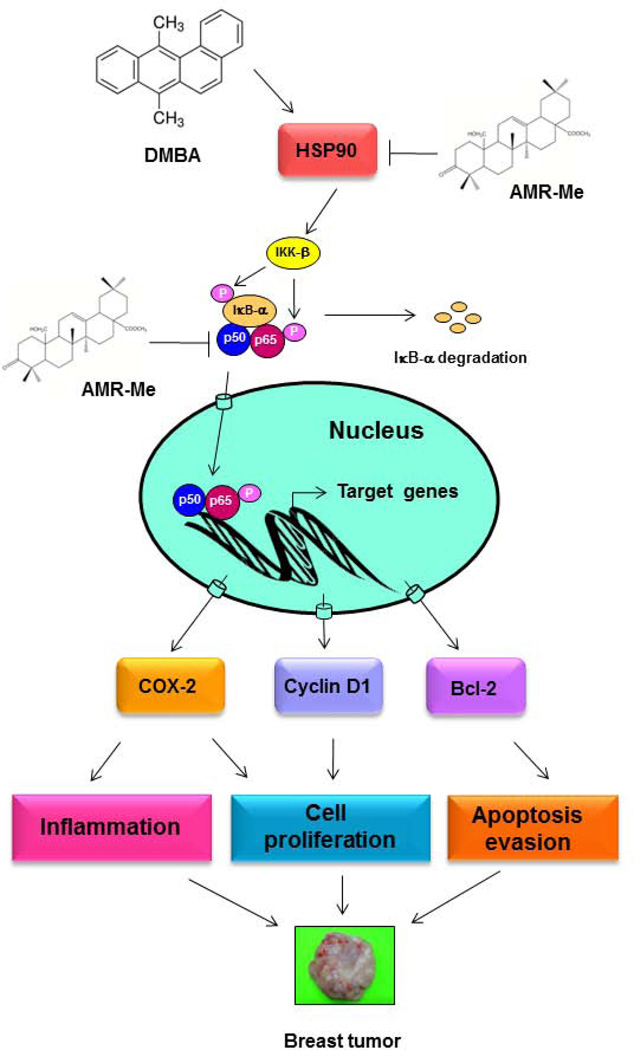
Breast cancer represents the second leading cause of cancer-related deaths among women worldwide and preventive therapy could reverse or delay the devastating impact of this disease. Methyl-amooranin (methyl-25-hydroxy-3-oxoolean-12-en-28-oate, AMR-Me), a novel synthetic oleanane triterpenoid, reduced the incidence and burden of 7,12-dimethylbenz(a)anthracene (DMBA)-induced mammary tumors in rats through antiproliferative and proapoptotic effects. Since chronic inflammation plays an important role in the pathogenesis of breast cancer and several synthetic oleanane compounds are known potent anti-inflammatory agents, we aim to investigate anti-inflammatory mechanisms of AMR-Me by monitoring various proinflammatory and stress markers, such as cyclooxygenase-2 (COX-2) and heat shock protein 90 (HSP90), and nuclear factor-κB (NF-κB) signaling during DMBA mammary tumorigenesis in rats. Mammary tumors were harvested from a chemopreventive study in which AMR-Me (0.8-1.6 mg/kg) was found to inhibit mammary carcinogenesis in a dose-response manner. The expressions of COX-2, HSP90, NF-κB, and inhibitory κB-α (IκB-α) were determined by immunohistochemistry and reverse transcription-polymerase chain reaction. AMR-Me downregulated the expression of intratumor COX-2 and HSP90, suppressed the degradation of IκB-α, and reduced the translocation of NF-κB from cytosol to nucleus. Our present study provides the first in vivo evidence that NF-κB-evoked inflammatory cascade is a major target of AMR-Me in breast cancer. Our current results together with our previous findings suggest that disruption of NF-κB signaling contributes to anti-inflammatory, antiproliferative, and apoptosis-inducing mechanisms involved in AMR-Me-mediated chemoprevention of rat mammary carcinogenesis. These encouraging mechanistic results coupled with a safety profile should facilitate the clinical development of AMR-Me as breast cancer chemopreventive drug.
Keywords: COX-2; DMBA; HSP90; NF-κB; breast tumor; inflammation; oleanane triterpenoid.
© 2013 Wiley Periodicals, Inc.
Figures
Similar articles
-
Simultaneous disruption of estrogen receptor and Wnt/β-catenin signaling is involved in methyl amooranin-mediated chemoprevention of mammary gland carcinogenesis in rats.Mol Cell Biochem. 2013 Dec;384(1-2):239-50. doi: 10.1007/s11010-013-1803-7. Mol Cell Biochem. 2013. PMID: 24078029 Free PMC article.
-
Anti-Inflammatory Mechanism Involved in Pomegranate-Mediated Prevention of Breast Cancer: the Role of NF-κB and Nrf2 Signaling Pathways.Nutrients. 2017 Apr 28;9(5):436. doi: 10.3390/nu9050436. Nutrients. 2017. PMID: 28452959 Free PMC article.
-
Chemopreventive effect of a novel oleanane triterpenoid in a chemically induced rodent model of breast cancer.Int J Cancer. 2013 Sep 1;133(5):1054-63. doi: 10.1002/ijc.28108. Epub 2013 Mar 29. Int J Cancer. 2013. PMID: 23404339 Free PMC article.
-
Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation.Mutat Res. 2001 Sep 1;480-481:243-68. doi: 10.1016/s0027-5107(01)00183-x. Mutat Res. 2001. PMID: 11506818 Review.
-
Chemoprevention of head and neck squamous cell carcinoma through inhibition of NF-κB signaling.Oral Oncol. 2014 Oct;50(10):930-41. doi: 10.1016/j.oraloncology.2013.10.005. Epub 2013 Oct 28. Oral Oncol. 2014. PMID: 24177052 Free PMC article. Review.
Cited by
-
Simultaneous disruption of estrogen receptor and Wnt/β-catenin signaling is involved in methyl amooranin-mediated chemoprevention of mammary gland carcinogenesis in rats.Mol Cell Biochem. 2013 Dec;384(1-2):239-50. doi: 10.1007/s11010-013-1803-7. Mol Cell Biochem. 2013. PMID: 24078029 Free PMC article.
-
Anti-Inflammatory Mechanism Involved in Pomegranate-Mediated Prevention of Breast Cancer: the Role of NF-κB and Nrf2 Signaling Pathways.Nutrients. 2017 Apr 28;9(5):436. doi: 10.3390/nu9050436. Nutrients. 2017. PMID: 28452959 Free PMC article.
-
Therapeutic and pharmacological efficacy of plant-derived bioactive compounds in targeting breast cancer.Am J Transl Res. 2024 May 15;16(5):1499-1520. doi: 10.62347/NUZN4999. eCollection 2024. Am J Transl Res. 2024. PMID: 38883353 Free PMC article. Review.
-
Trianthema portulacastrum Linn. displays anti-inflammatory responses during chemically induced rat mammary tumorigenesis through simultaneous and differential regulation of NF-κB and Nrf2 signaling pathways.Int J Mol Sci. 2015 Jan 22;16(2):2426-45. doi: 10.3390/ijms16022426. Int J Mol Sci. 2015. PMID: 25622256 Free PMC article.
-
Oleanolic acid and its synthetic derivatives for the prevention and therapy of cancer: preclinical and clinical evidence.Cancer Lett. 2014 May 1;346(2):206-16. doi: 10.1016/j.canlet.2014.01.016. Epub 2014 Jan 30. Cancer Lett. 2014. PMID: 24486850 Free PMC article. Review.
References
-
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. - PubMed
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–33. - PubMed
-
- Crujeiras AB, Díaz-Lagares A, Carreira MC, Amil M, Casanueva FF. Oxidative stress associated to dysfunctional adipose tissue: a potential link between obesity, type 2 diabetes mellitus and breast cancer. Free Radic Res. 2013 in press. PMID: 23409968. - PubMed
-
- Pierobon M, Frankenfeld CL. Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013;137:307–314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
