

Molecular mechanisms of cognitive dysfunction following traumatic brain injury
- PMID: 23847533
- PMCID: PMC3705200
- DOI: 10.3389/fnagi.2013.00029
Molecular mechanisms of cognitive dysfunction following traumatic brain injury
Abstract
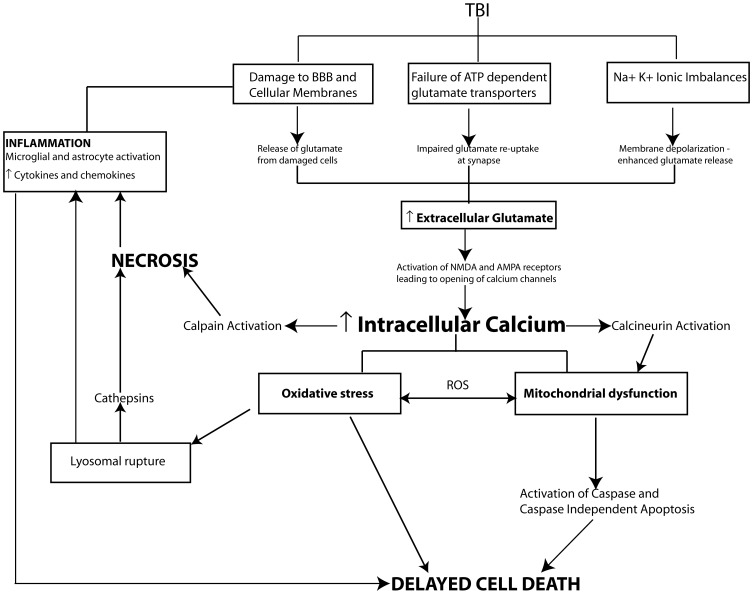
Traumatic brain injury (TBI) results in significant disability due to cognitive deficits particularly in attention, learning and memory, and higher-order executive functions. The role of TBI in chronic neurodegeneration and the development of neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), Amyotrophic Lateral Sclerosis (ALS) and most recently chronic traumatic encephalopathy (CTE) is of particular importance. However, despite significant effort very few therapeutic options exist to prevent or reverse cognitive impairment following TBI. In this review, we present experimental evidence of the known secondary injury mechanisms which contribute to neuronal cell loss, axonal injury, and synaptic dysfunction and hence cognitive impairment both acutely and chronically following TBI. In particular we focus on the mechanisms linking TBI to the development of two forms of dementia: AD and CTE. We provide evidence of potential molecular mechanisms involved in modulating Aβ and Tau following TBI and provide evidence of the role of these mechanisms in AD pathology. Additionally we propose a mechanism by which Aβ generated as a direct result of TBI is capable of exacerbating secondary injury mechanisms thereby establishing a neurotoxic cascade that leads to chronic neurodegeneration.
Keywords: Alzheimer's; Aβ; CTE; TBI; cognition; tau.
Figures




References
-
- Abrahamson E. E., Ikonomovic M. D., Ciallella J. R., Hope C. E., Paljug W. R., Isanski B. A., et al. (2006). Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp. Neurol. 197, 437–450 10.1016/j.expneurol.2005.10.011 - DOI - PubMed
-
- Acosta S. A., Tajiri N., Shinozuka K., Ishikawa H., Grimmig B., Diamond D., et al. (2013). Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 8:e53376 10.1371/journal.pone.0053376 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous