

Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3
- PMID: 23848338
- PMCID: PMC3799595
- DOI: 10.1111/bph.12302
Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3
Abstract
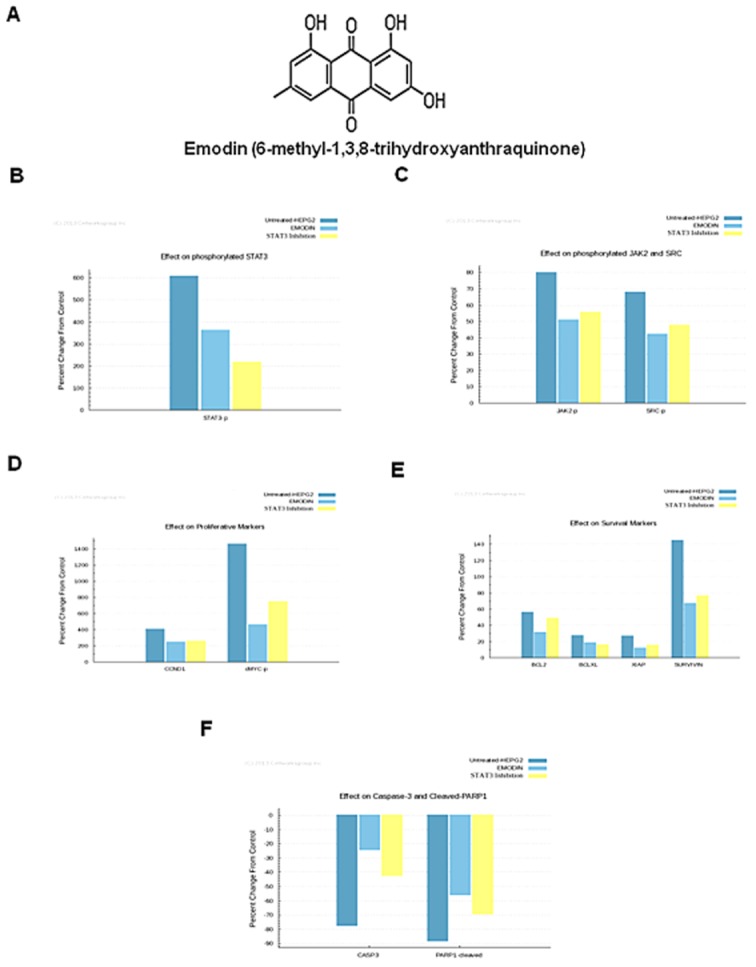
Background and purpose: Aberrant activation of STAT3 is frequently encountered and promotes proliferation, survival, metastasis and angiogenesis in hepatocellular carcinoma (HCC). Here, we have investigated whether emodin mediates its effect through interference with the STAT3 activation pathway in HCC.
Experimental approach: The effect of emodin on STAT3 activation, associated protein kinases and apoptosis was investigated using various HCC cell lines. Additionally, we also used a predictive tumour technology to analyse the effects of emodin . The in vivo effects of emodin were assessed in an orthotopic mouse model of HCC.
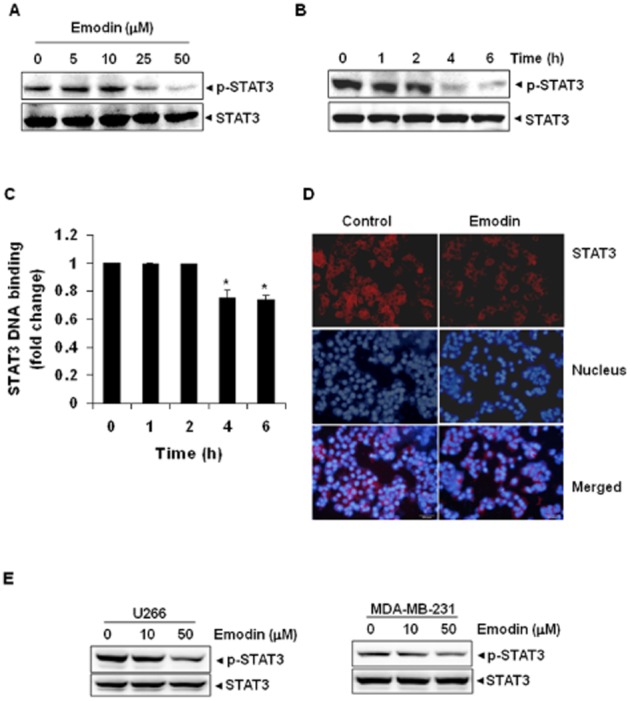
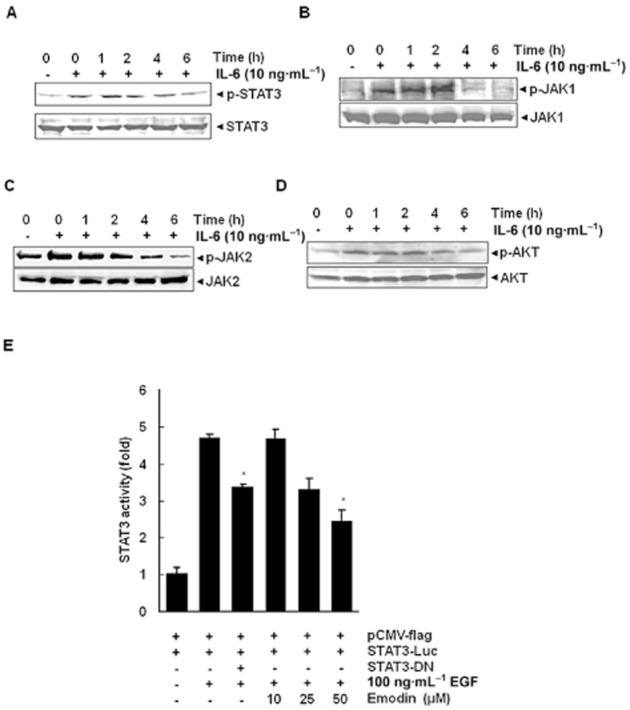
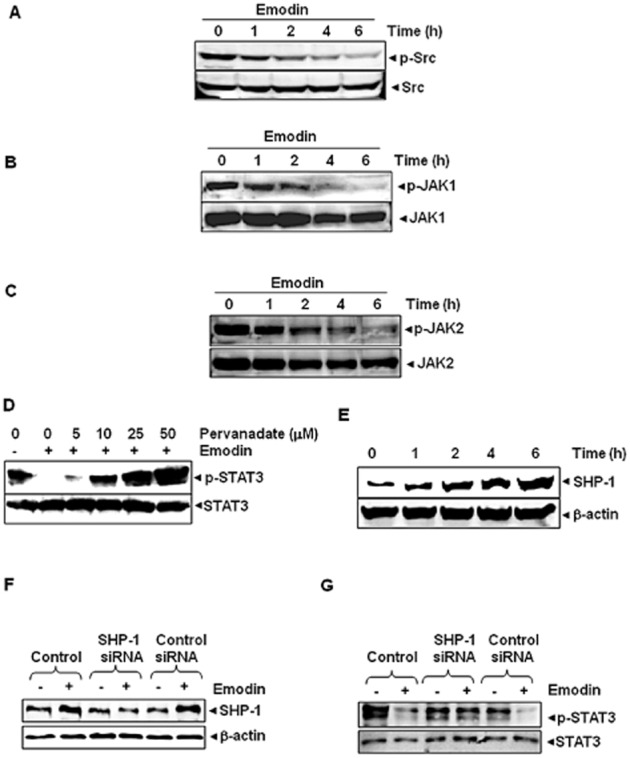
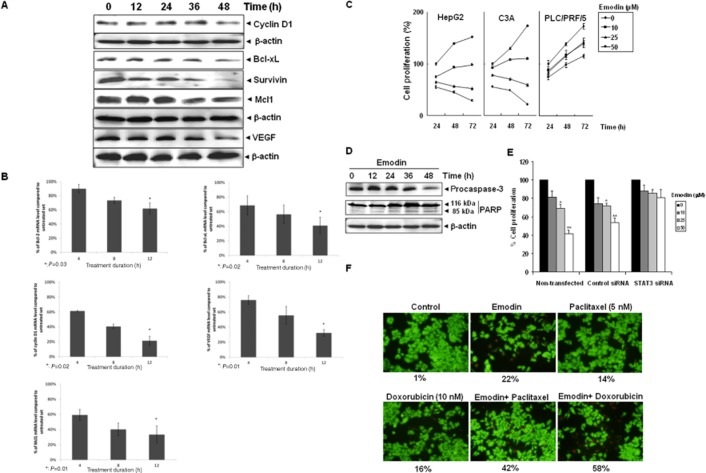
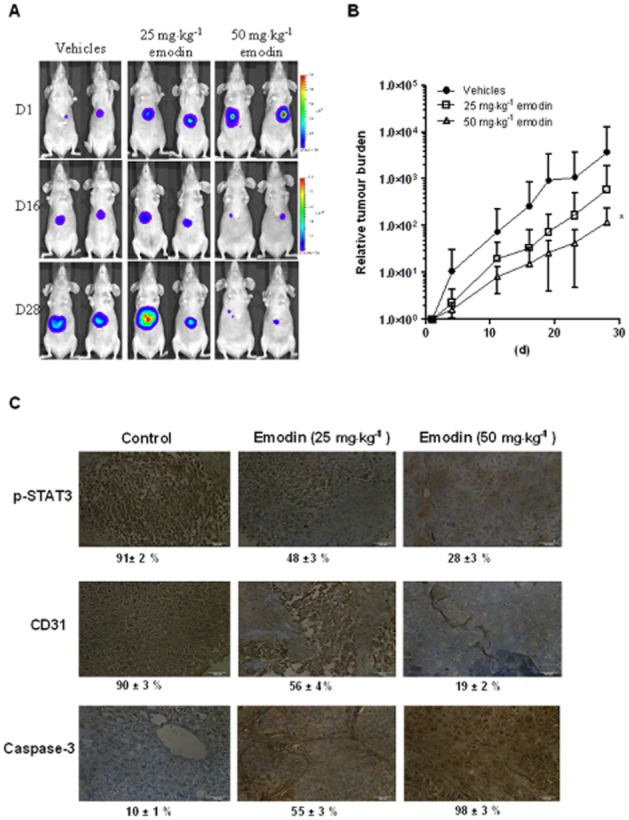
Key results: Emodin suppressed STAT3 activation in a dose- and time-dependent manner in HCC cells, mediated by the modulation of activation of upstream kinases c-Src, JAK1 and JAK2. Vanadate treatment reversed emodin-induced down-regulation of STAT3, suggesting the involvement of a tyrosine phosphatase and emodin induced the expression of the tyrosine phosphatase SHP-1 that correlated with the down-regulation of constitutive STAT3 activation. Interestingly, silencing of the SHP-1 gene by siRNA abolished the ability of emodin to inhibit STAT3 activation. Finally, when administered i.p., emodin inhibited the growth of human HCC orthotopic tumours in male athymic nu/nu mice and STAT3 activation in tumour tissues.
Conclusions and implications: Emodin mediated its effects predominantly through inhibition of the STAT3 signalling cascade and thus has a particular potential for the treatment of cancers expressing constitutively activated STAT3.
Keywords: STAT3; apoptosis; emodin; hepatocellular carcinoma; proliferation.
© 2013 The British Pharmacological Society.
Figures
References
-
- Battistutta R, Sarno S, De Moliner E, Papinutto E, Zanotti G, Pinna LA. The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J Biol Chem. 2000;275:29618–29622. - PubMed
-
- Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–1128. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
