

Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology
- PMID: 23852728
- PMCID: PMC3732980
- DOI: 10.1073/pnas.1302455110
Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology
Abstract
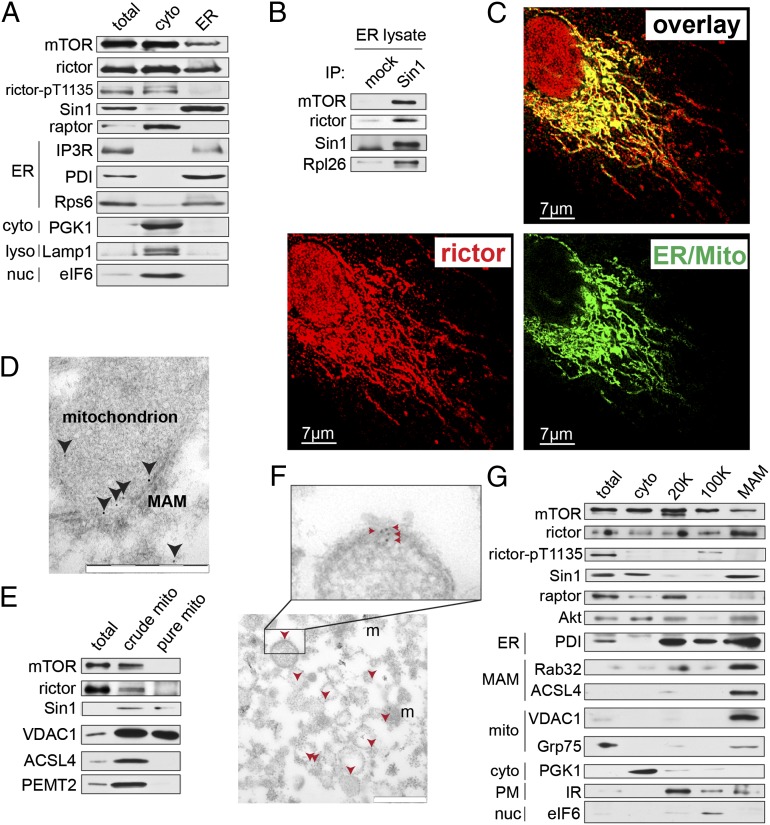
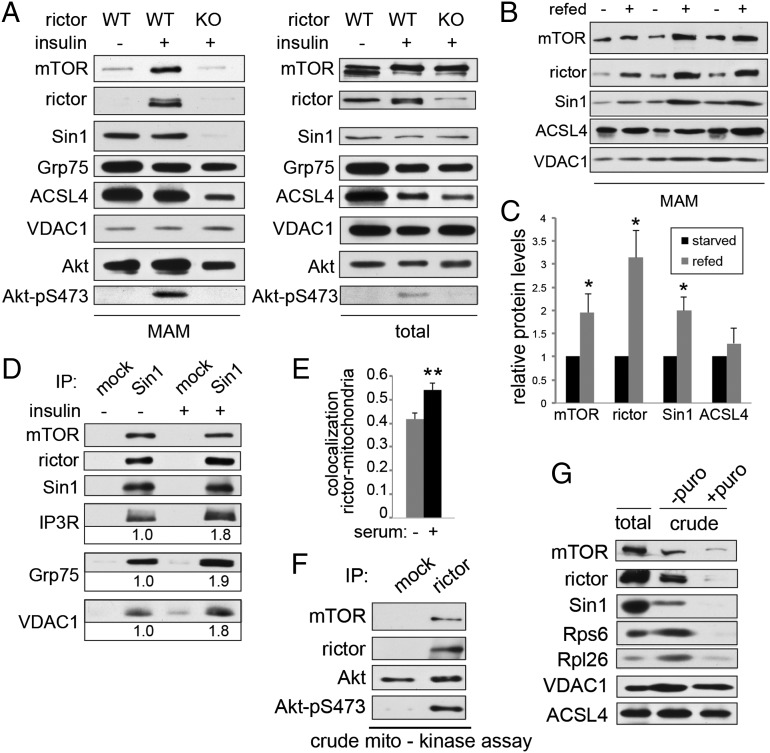
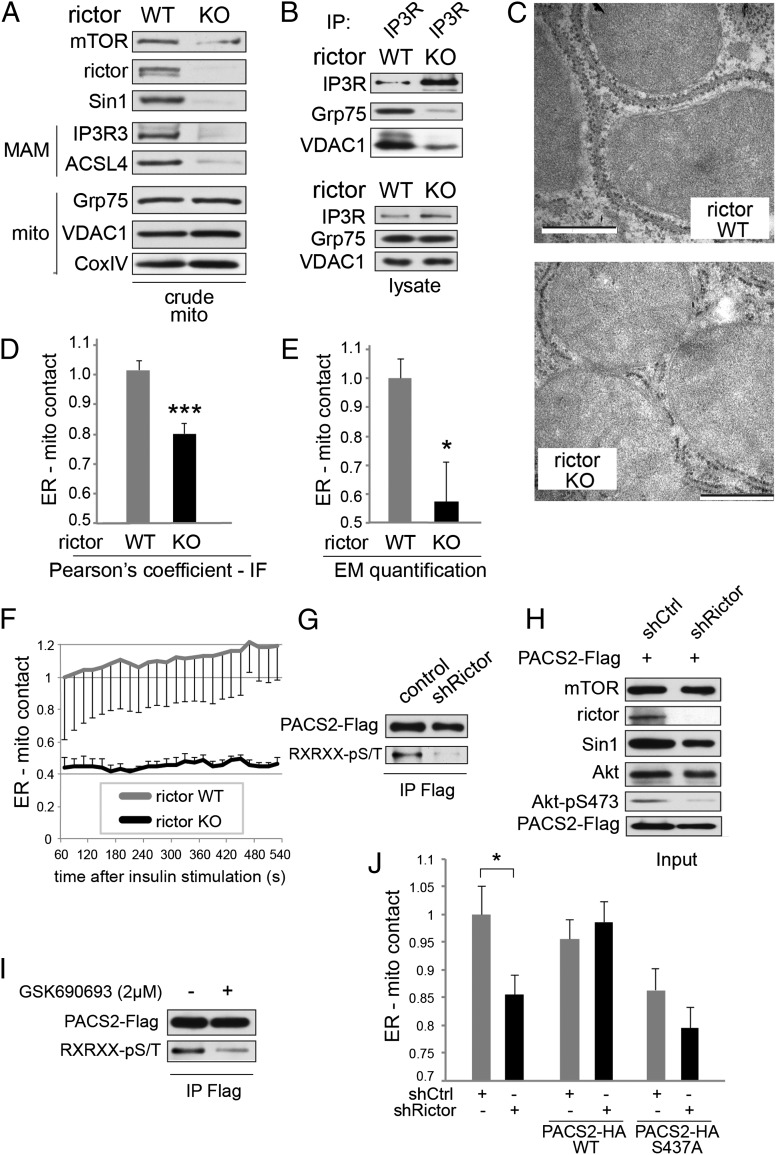
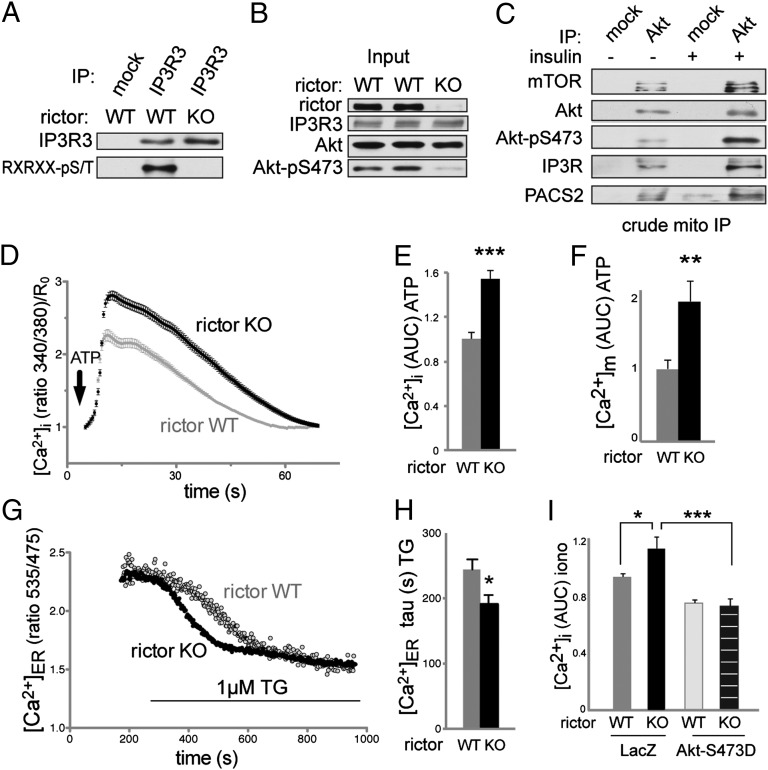
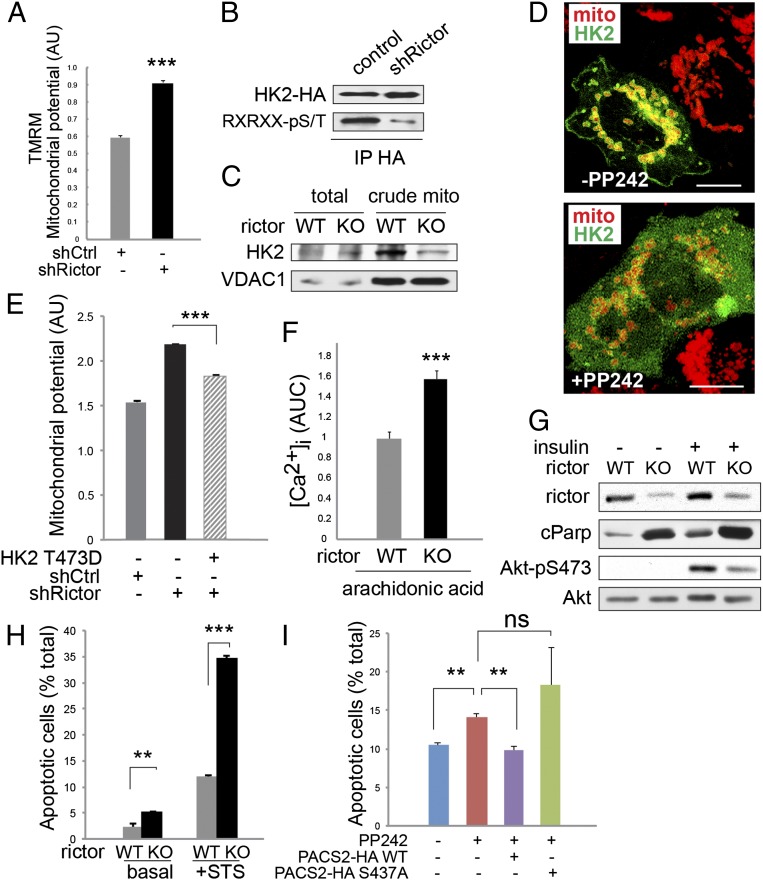
The target of rapamycin (TOR) is a highly conserved protein kinase and a central controller of growth. Mammalian TOR complex 2 (mTORC2) regulates AGC kinase family members and is implicated in various disorders, including cancer and diabetes. Here we report that mTORC2 is localized to the endoplasmic reticulum (ER) subcompartment termed mitochondria-associated ER membrane (MAM). mTORC2 localization to MAM was growth factor-stimulated, and mTORC2 at MAM interacted with the IP3 receptor (IP3R)-Grp75-voltage-dependent anion-selective channel 1 ER-mitochondrial tethering complex. mTORC2 deficiency disrupted MAM, causing mitochondrial defects including increases in mitochondrial membrane potential, ATP production, and calcium uptake. mTORC2 controlled MAM integrity and mitochondrial function via Akt mediated phosphorylation of the MAM associated proteins IP3R, Hexokinase 2, and phosphofurin acidic cluster sorting protein 2. Thus, mTORC2 is at the core of a MAM signaling hub that controls growth and metabolism.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Rizzuto R, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280(5370):1763–1766. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
