

DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis
- PMID: 23853579
- PMCID: PMC3701705
- DOI: 10.1371/journal.ppat.1003419
DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis
Abstract
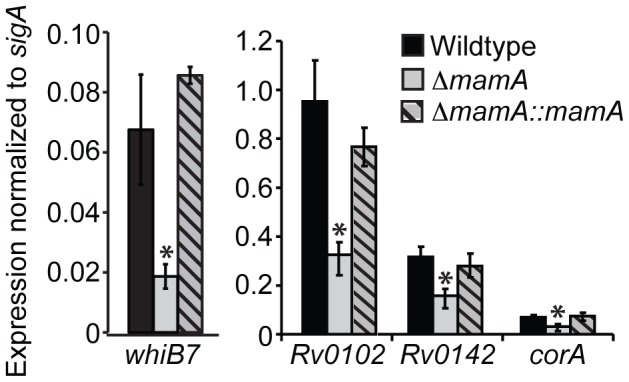
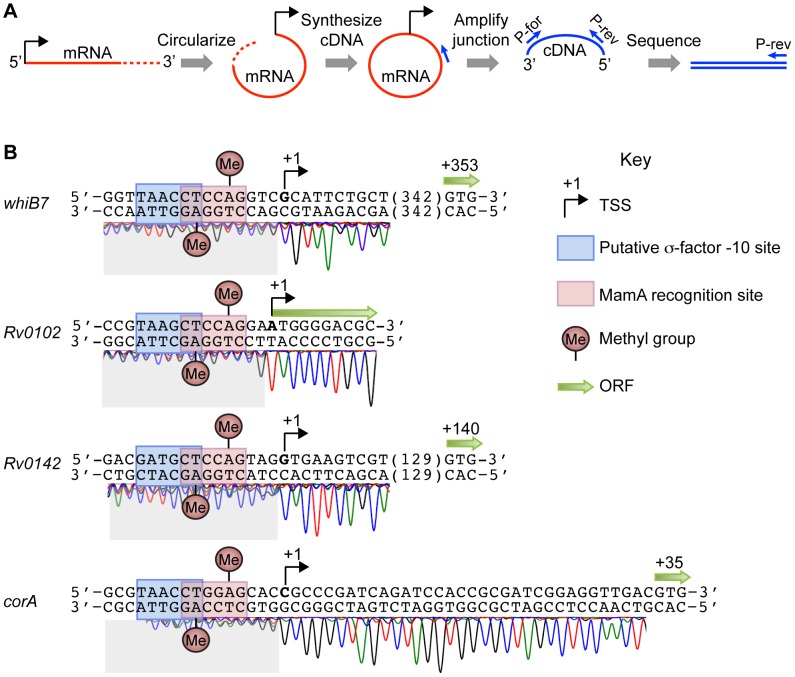
DNA methylation regulates gene expression in many organisms. In eukaryotes, DNA methylation is associated with gene repression, while it exerts both activating and repressive effects in the Proteobacteria through largely locus-specific mechanisms. Here, we identify a critical DNA methyltransferase in M. tuberculosis, which we term MamA. MamA creates N⁶-methyladenine in a six base pair recognition sequence present in approximately 2,000 copies on each strand of the genome. Loss of MamA reduces the expression of a number of genes. Each has a MamA site located at a conserved position relative to the sigma factor -10 binding site and transcriptional start site, suggesting that MamA modulates their expression through a shared, not locus-specific, mechanism. While strains lacking MamA grow normally in vitro, they are attenuated in hypoxic conditions, suggesting that methylation promotes survival in discrete host microenvironments. Interestingly, we demonstrate strikingly different patterns of DNA methyltransferase activity in different lineages of M. tuberculosis, which have been associated with preferences for distinct host environments and different disease courses in humans. Thus, MamA is the major functional adenine methyltransferase in M. tuberculosis strains of the Euro-American lineage while strains of the Beijing lineage harbor a point mutation that largely inactivates MamA but possess a second functional DNA methyltransferase. Our results indicate that MamA influences gene expression in M. tuberculosis and plays an important but strain-specific role in fitness during hypoxia.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- World Health Organization (2011) Global Tuberculosis Control 2011. World Health Organization.
-
- Militello KT, Simon RD, Qureshi M, Maines R, VanHorne ML, et al. (2012) Conservation of Dcm-mediated cytosine DNA methylation in Escherichia coli. FEMS Microbiology Letters 328: 78–85 doi:10.1111/j.1574-6968.2011.02482.x - DOI - PMC - PubMed
-
- Kahramanoglou C, Prieto AI, Khedkar S, Haase B, Gupta A, et al. (2012) Genomics of DNA cytosine methylation in Escherichia coli reveals its role in stationary phase transcription. Nat Commun 3: 886 doi:10.1038/ncomms1878 - DOI - PubMed
-
- Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics 9: 465–476 doi:10.1038/nrg2341 - DOI - PubMed
-
- Casadesus J, Low D (2006) Epigenetic Gene Regulation in the Bacterial World. Microbiology and Molecular Biology Reviews 70: 830–856 doi:10.1128/MMBR.00016-06 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
