

Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology
- PMID: 23857503
- PMCID: PMC4001082
- DOI: 10.1001/jamainternmed.2013.7734
Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology
Abstract
Importance: The latest generation of benchtop DNA sequencing platforms can provide an accurate whole-genome sequence (WGS) for a broad range of bacteria in less than a day. These could be used to more effectively contain the spread of multidrug-resistant pathogens.
Objective: To compare WGS with standard clinical microbiology practice for the investigation of nosocomial outbreaks caused by multidrug-resistant bacteria, the identification of genetic determinants of antimicrobial resistance, and typing of other clinically important pathogens.
Design, setting, and participants: A laboratory-based study of hospital inpatients with a range of bacterial infections at Cambridge University Hospitals NHS Foundation Trust, a secondary and tertiary referral center in England, comparing WGS with standard diagnostic microbiology using stored bacterial isolates and clinical information.
Main outcomes and measures: Specimens were taken and processed as part of routine clinical care, and cultured isolates stored and referred for additional reference laboratory testing as necessary. Isolates underwent DNA extraction and library preparation prior to sequencing on the Illumina MiSeq platform. Bioinformatic analyses were performed by persons blinded to the clinical, epidemiologic, and antimicrobial susceptibility data.
Results: We investigated 2 putative nosocomial outbreaks, one caused by vancomycin-resistant Enterococcus faecium and the other by carbapenem-resistant Enterobacter cloacae; WGS accurately discriminated between outbreak and nonoutbreak isolates and was superior to conventional typing methods. We compared WGS with standard methods for the identification of the mechanism of carbapenem resistance in a range of gram-negative bacteria (Acinetobacter baumannii, E cloacae, Escherichia coli, and Klebsiella pneumoniae). This demonstrated concordance between phenotypic and genotypic results, and the ability to determine whether resistance was attributable to the presence of carbapenemases or other resistance mechanisms. Whole-genome sequencing was used to recapitulate reference laboratory typing of clinical isolates of Neisseria meningitidis and to provide extended phylogenetic analyses of these.
Conclusions and relevance: The speed, accuracy, and depth of information provided by WGS platforms to confirm or refute outbreaks in hospitals and the community, and to accurately define transmission of multidrug-resistant and other organisms, represents an important advance.
Figures
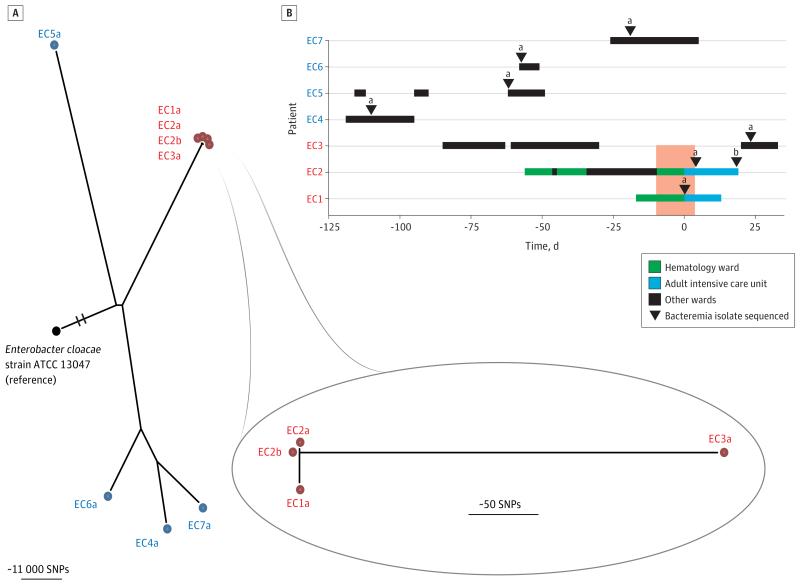
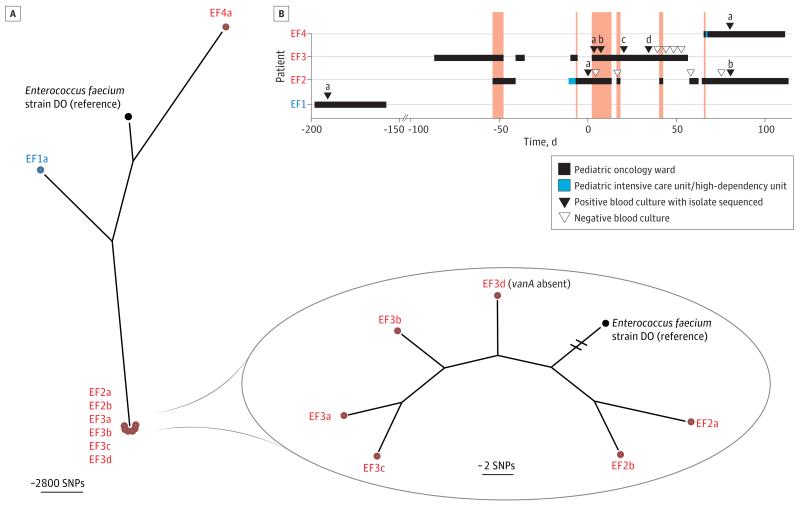
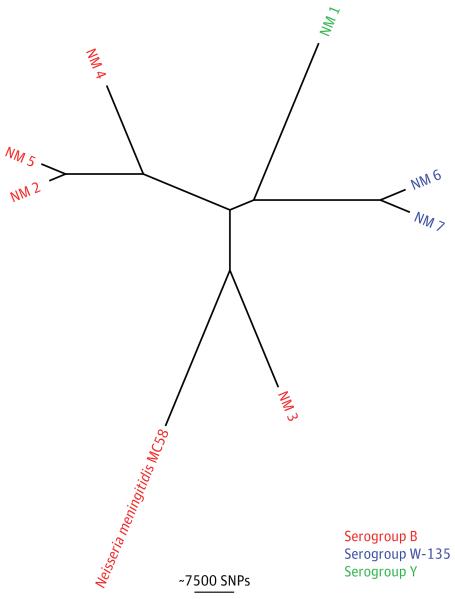
Comment in
-
The time is now for gene- and genome-based bacterial diagnostics: "you say you want a revolution".JAMA Intern Med. 2013 Aug 12;173(15):1405-6. doi: 10.1001/jamainternmed.2013.7042. JAMA Intern Med. 2013. PMID: 23857368 No abstract available.
References
-
- Gardy JL, Johnston JC, Ho Sui SJ, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364(8):730–739. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
