

Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and "fibroblast addiction" are new therapeutic targets for drug discovery
- PMID: 23860382
- PMCID: PMC3899185
- DOI: 10.4161/cc.25695
Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and "fibroblast addiction" are new therapeutic targets for drug discovery
Abstract
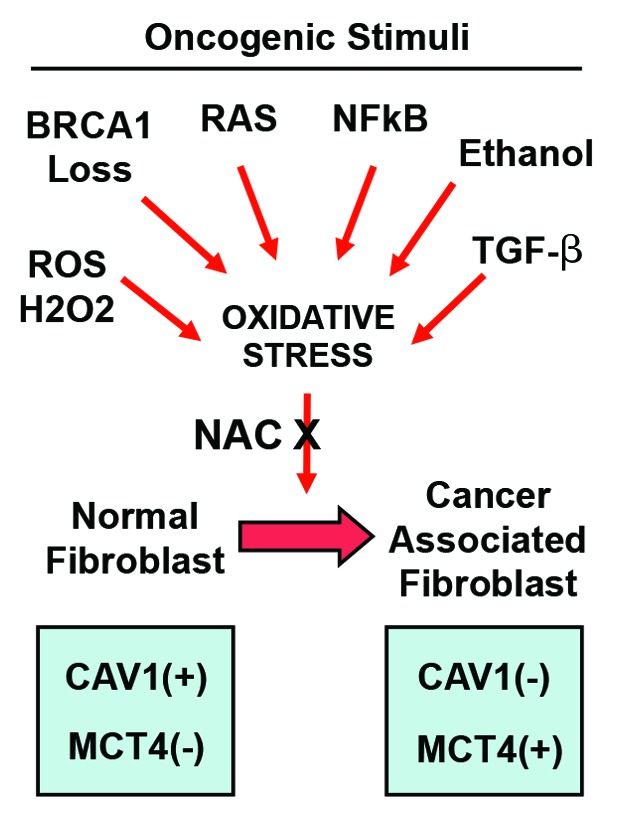
Metabolic coupling, between mitochondria in cancer cells and catabolism in stromal fibroblasts, promotes tumor growth, recurrence, metastasis, and predicts anticancer drug resistance. Catabolic fibroblasts donate the necessary fuels (such as L-lactate, ketones, glutamine, other amino acids, and fatty acids) to anabolic cancer cells, to metabolize via their TCA cycle and oxidative phosphorylation (OXPHOS). This provides a simple mechanism by which metabolic energy and biomass are transferred from the host microenvironment to cancer cells. Recently, we showed that catabolic metabolism and "glycolytic reprogramming" in the tumor microenvironment are orchestrated by oncogene activation and inflammation, which originates in epithelial cancer cells. Oncogenes drive the onset of the cancer-associated fibroblast phenotype in adjacent normal fibroblasts via paracrine oxidative stress. This oncogene-induced transition to malignancy is "mirrored" by a loss of caveolin-1 (Cav-1) and an increase in MCT4 in adjacent stromal fibroblasts, functionally reflecting catabolic metabolism in the tumor microenvironment. Virtually identical findings were obtained using BRCA1-deficient breast and ovarian cancer cells. Thus, oncogene activation (RAS, NFkB, TGF-β) and/or tumor suppressor loss (BRCA1) have similar functional effects on adjacent stromal fibroblasts, initiating "metabolic symbiosis" and the cancer-associated fibroblast phenotype. New therapeutic strategies that metabolically uncouple oxidative cancer cells from their glycolytic stroma or modulate oxidative stress could be used to target this lethal subtype of cancers. Targeting "fibroblast addiction" in primary and metastatic tumor cells may expose a critical Achilles' heel, leading to disease regression in both sporadic and familial cancers.
Keywords: BRCA1; NFkB; RAS; TGF-beta; cancer-associated fibroblast; glycolysis; metabolic symbiosis; oncogene; oxidative stress; stromal biomarkers; tumor microenvironment; tumor suppressor.
Figures








Similar articles
-
Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFκB target stromal MCT4.Cell Cycle. 2013 Aug 15;12(16):2580-97. doi: 10.4161/cc.25510. Epub 2013 Jul 8. Cell Cycle. 2013. PMID: 23860378 Free PMC article.
-
Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with "Warburg-like" cancer metabolism and L-lactate production.Cell Cycle. 2012 Aug 15;11(16):3019-35. doi: 10.4161/cc.21384. Epub 2012 Aug 9. Cell Cycle. 2012. PMID: 22874531 Free PMC article.
-
BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies.Cell Cycle. 2012 Dec 1;11(23):4402-13. doi: 10.4161/cc.22776. Epub 2012 Nov 21. Cell Cycle. 2012. PMID: 23172369 Free PMC article.
-
Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth.Semin Cancer Biol. 2014 Apr;25:47-60. doi: 10.1016/j.semcancer.2014.01.005. Epub 2014 Jan 28. Semin Cancer Biol. 2014. PMID: 24486645 Review.
-
Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function.Semin Oncol. 2014 Apr;41(2):195-216. doi: 10.1053/j.seminoncol.2014.03.002. Epub 2014 Mar 5. Semin Oncol. 2014. PMID: 24787293 Review.
Cited by
-
Recent advances in understanding the metabolic plasticity of ovarian cancer: A systematic review.Heliyon. 2022 Nov 11;8(11):e11487. doi: 10.1016/j.heliyon.2022.e11487. eCollection 2022 Nov. Heliyon. 2022. PMID: 36406733 Free PMC article.
-
Energy metabolism in cancer stem cells.World J Stem Cells. 2020 Jun 26;12(6):448-461. doi: 10.4252/wjsc.v12.i6.448. World J Stem Cells. 2020. PMID: 32742562 Free PMC article. Review.
-
Metabolic Interactions Between Tumor and Stromal Cells in the Tumor Microenvironment.Adv Exp Med Biol. 2021;1350:101-121. doi: 10.1007/978-3-030-83282-7_5. Adv Exp Med Biol. 2021. PMID: 34888846
-
Clinicopathological and prognostic significance of caveolin-1 and ATG4C expression in the epithelial ovarian cancer.PLoS One. 2020 May 13;15(5):e0232235. doi: 10.1371/journal.pone.0232235. eCollection 2020. PLoS One. 2020. PMID: 32401768 Free PMC article.
-
Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells.Nat Cell Biol. 2018 May;20(5):597-609. doi: 10.1038/s41556-018-0083-6. Epub 2018 Apr 16. Nat Cell Biol. 2018. PMID: 29662176 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous