

Mutations in DSTYK and dominant urinary tract malformations
- PMID: 23862974
- PMCID: PMC3846391
- DOI: 10.1056/NEJMoa1214479
Mutations in DSTYK and dominant urinary tract malformations
Abstract
Background: Congenital abnormalities of the kidney and the urinary tract are the most common cause of pediatric kidney failure. These disorders are highly heterogeneous, and the etiologic factors are poorly understood.
Methods: We performed genomewide linkage analysis and whole-exome sequencing in a family with an autosomal dominant form of congenital abnormalities of the kidney or urinary tract (seven affected family members). We also performed a sequence analysis in 311 unrelated patients, as well as histologic and functional studies.
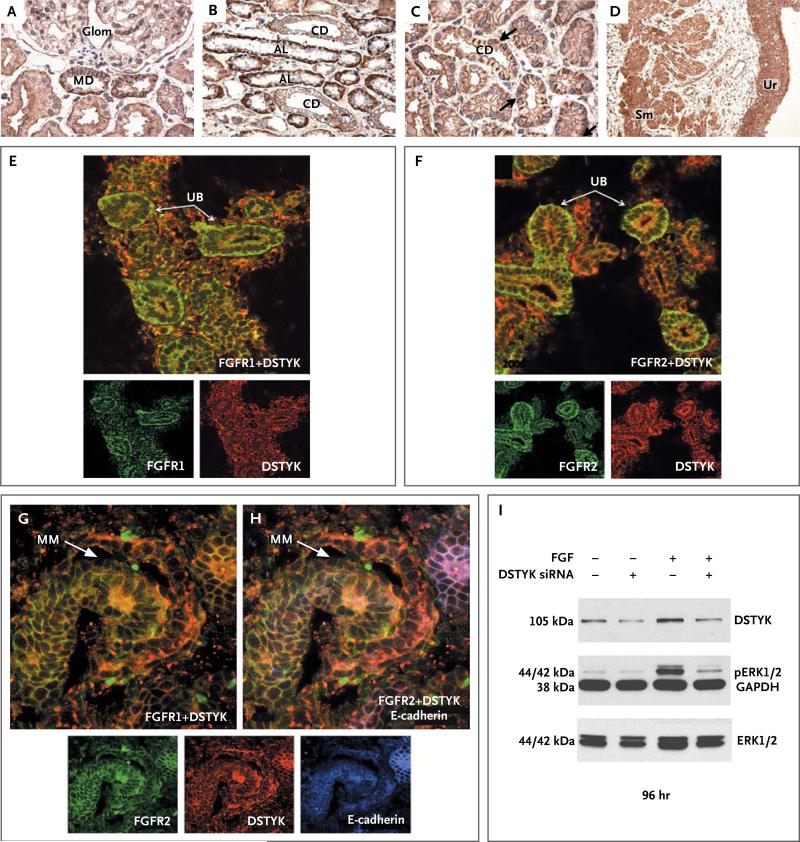
Results: Linkage analysis identified five regions of the genome that were shared among all affected family members. Exome sequencing identified a single, rare, deleterious variant within these linkage intervals, a heterozygous splice-site mutation in the dual serine-threonine and tyrosine protein kinase gene (DSTYK). This variant, which resulted in aberrant splicing of messenger RNA, was present in all affected family members. Additional, independent DSTYK mutations, including nonsense and splice-site mutations, were detected in 7 of 311 unrelated patients. DSTYK is highly expressed in the maturing epithelia of all major organs, localizing to cell membranes. Knockdown in zebrafish resulted in developmental defects in multiple organs, which suggested loss of fibroblast growth factor (FGF) signaling. Consistent with this finding is the observation that DSTYK colocalizes with FGF receptors in the ureteric bud and metanephric mesenchyme. DSTYK knockdown in human embryonic kidney cells inhibited FGF-stimulated phosphorylation of extracellular-signal-regulated kinase (ERK), the principal signal downstream of receptor tyrosine kinases.
Conclusions: We detected independent DSTYK mutations in 2.3% of patients with congenital abnormalities of the kidney or urinary tract, a finding that suggests that DSTYK is a major determinant of human urinary tract development, downstream of FGF signaling. (Funded by the National Institutes of Health and others.).
Figures

Comment in
-
Genetics: DSTYK gene linked to urinary tract defects.Nat Rev Urol. 2013 Sep;10(9):492. doi: 10.1038/nrurol.2013.171. Epub 2013 Aug 6. Nat Rev Urol. 2013. PMID: 23917117 No abstract available.
-
Genetics and urinary tract malformations.Am J Kidney Dis. 2014 Feb;63(2):183-5. doi: 10.1053/j.ajkd.2013.11.005. Epub 2013 Dec 4. Am J Kidney Dis. 2014. PMID: 24315771 No abstract available.
References
-
- Loane M, Dolk H, Kelly A, Teljeur C, Greenlees R, Densem J. Paper 4: EUROCAT statistical monitoring: identification and investigation of ten year trends of congenital anomalies in Europe. Birth Defects Res A Clin Mol Teratol. 2011;91(Suppl 1):S31–S43. - PubMed
-
- Birth Defects Monitoring Program (BDMP)/Commission on Professional and Hospital Activities (CPHA) surveillance data, 1988–1991. Teratology. 1993;48:658–75. - PubMed
-
- Ardissino G, Dacco V, Testa S, et al. Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics. 2003;111:e382–e387. - PubMed
-
- Sanna-Cherchi S, Ravani P, Corbani V, et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009;76:528–33. - PubMed
-
- Woolf AS, Winyard PJ. Molecular mechanisms of human embryogenesis: developmental pathogenesis of renal tract malformations. Pediatr Dev Pathol. 2002;5:108–29. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K23-DK090207/DK/NIDDK NIH HHS/United States
- UM1 HG006504/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- K23 DK090207/DK/NIDDK NIH HHS/United States
- DK071041/DK/NIDDK NIH HHS/United States
- P01 DK055388/DK/NIDDK NIH HHS/United States
- U54 HG006504/HG/NHGRI NIH HHS/United States
- K08 DK078014/DK/NIDDK NIH HHS/United States
- R01 DK053093/DK/NIDDK NIH HHS/United States
- R01 DK080099/DK/NIDDK NIH HHS/United States
- GGP08050/TI_/Telethon/Italy
- HG006504/HG/NHGRI NIH HHS/United States
- R01 DK071041/DK/NIDDK NIH HHS/United States
- P30 DK079310/DK/NIDDK NIH HHS/United States
- 1R01DK080099/DK/NIDDK NIH HHS/United States
- R01 DK020999/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous