

Rap-afadin axis in control of Rho signaling and endothelial barrier recovery
- PMID: 23864716
- PMCID: PMC3756920
- DOI: 10.1091/mbc.E13-02-0098
Rap-afadin axis in control of Rho signaling and endothelial barrier recovery
Abstract
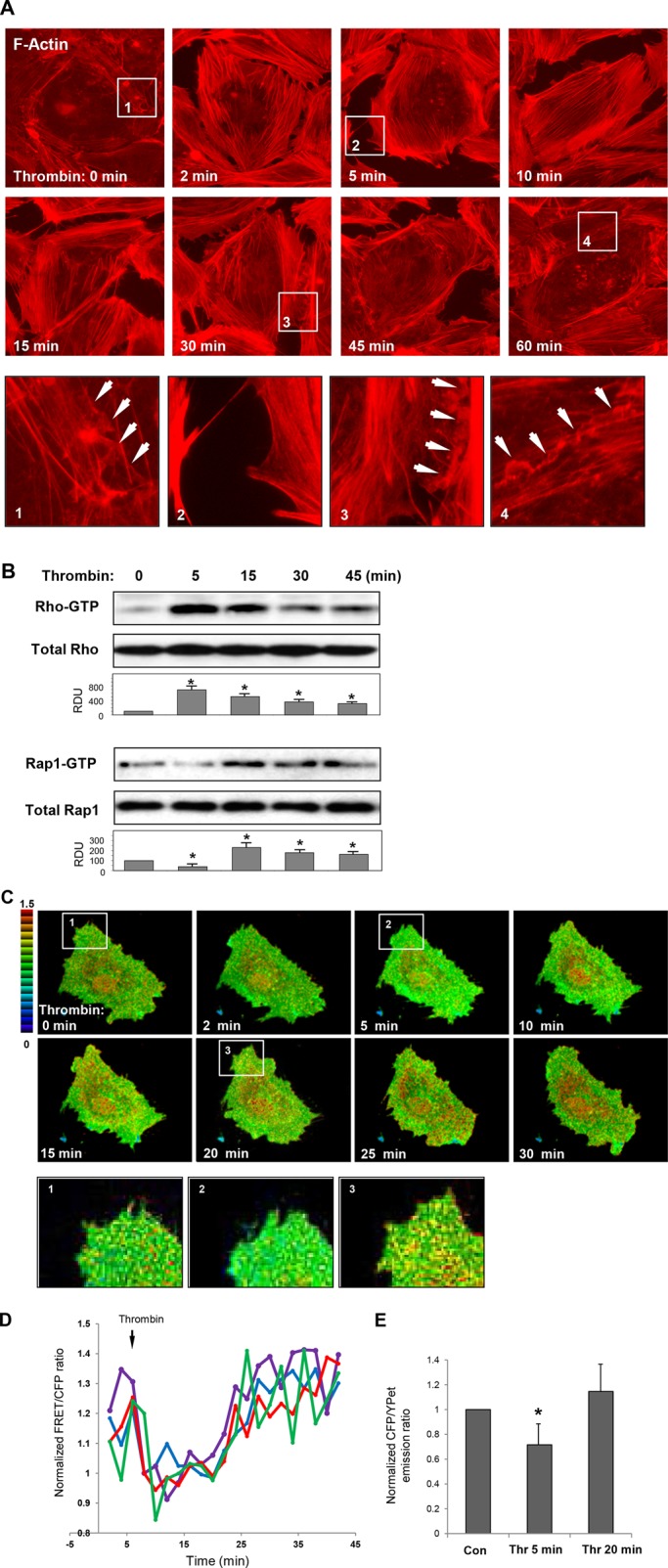
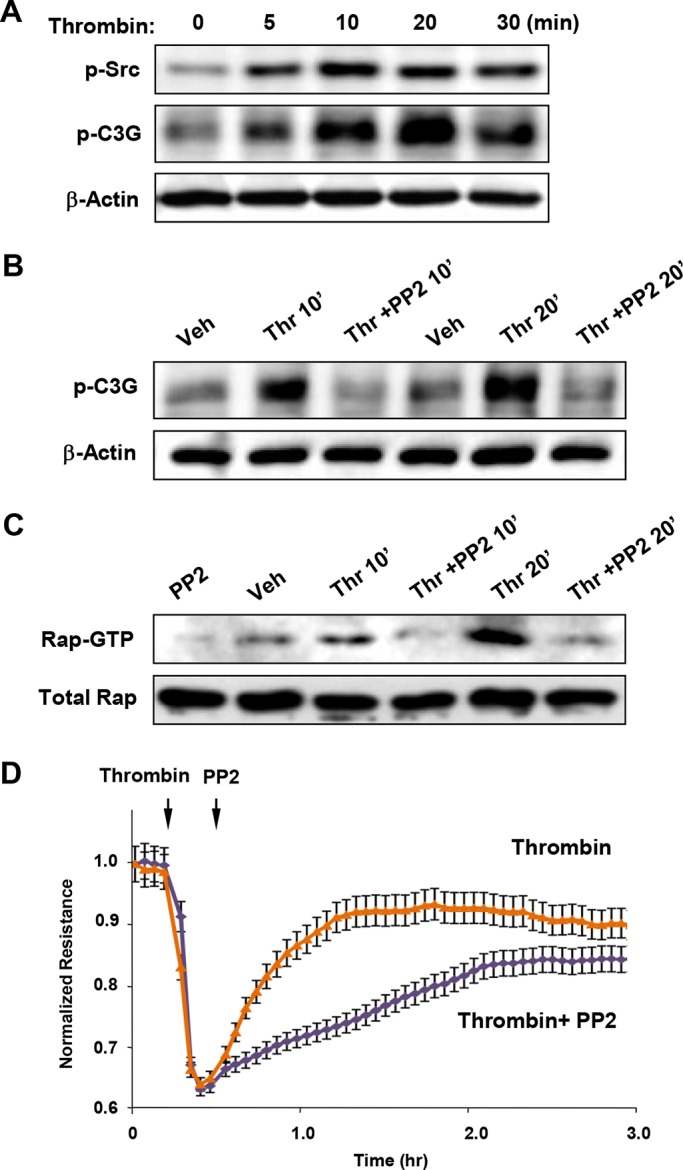
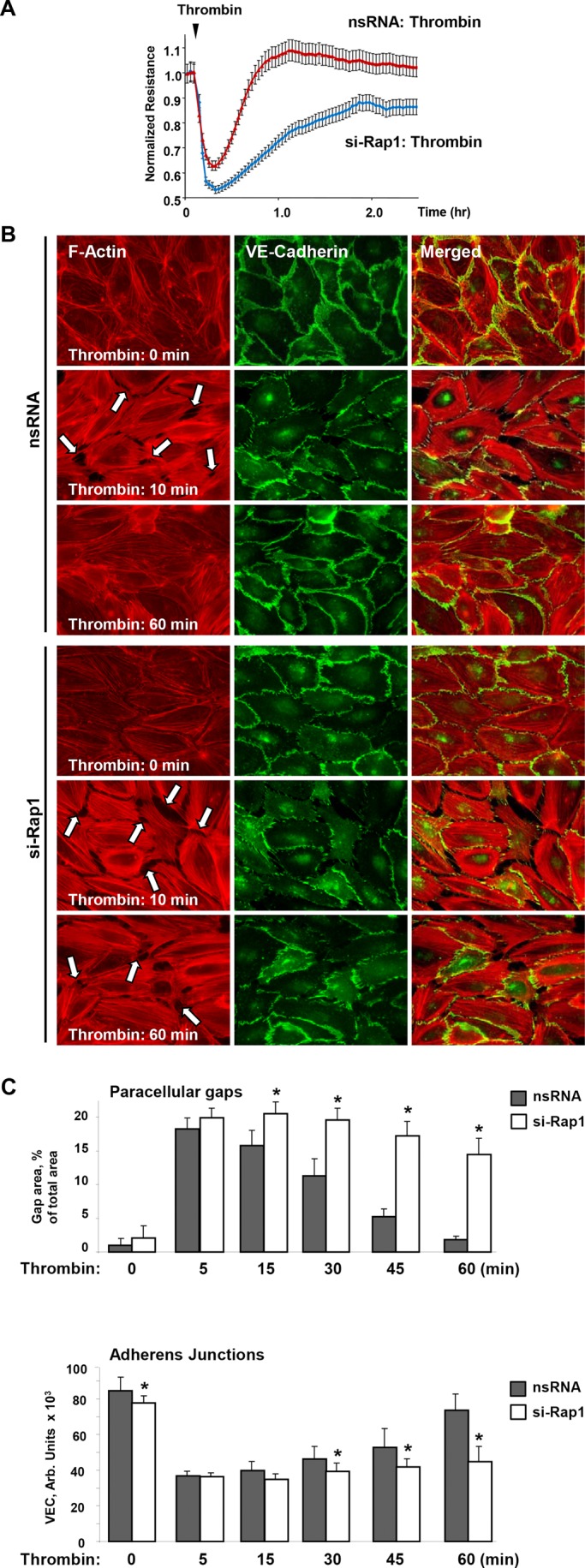
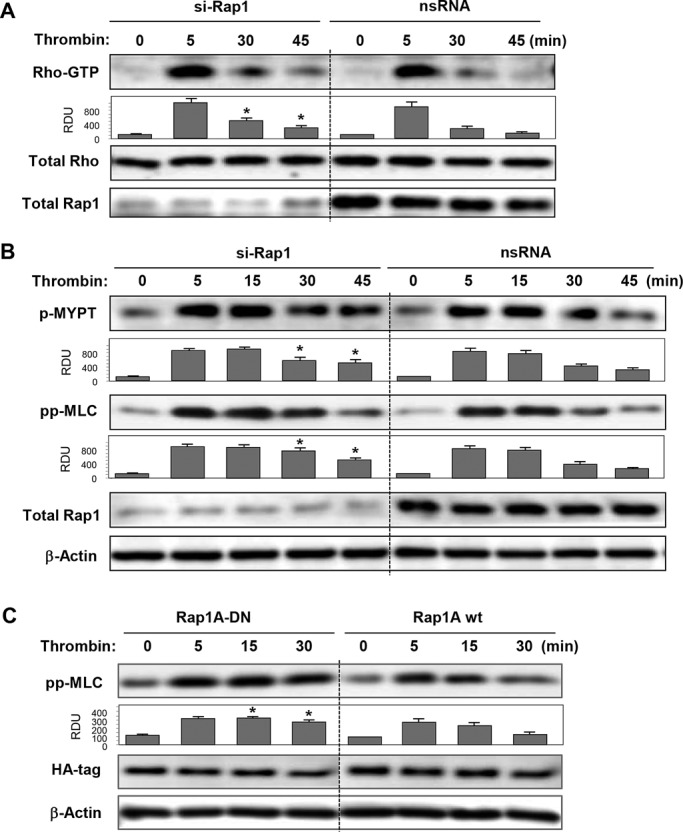
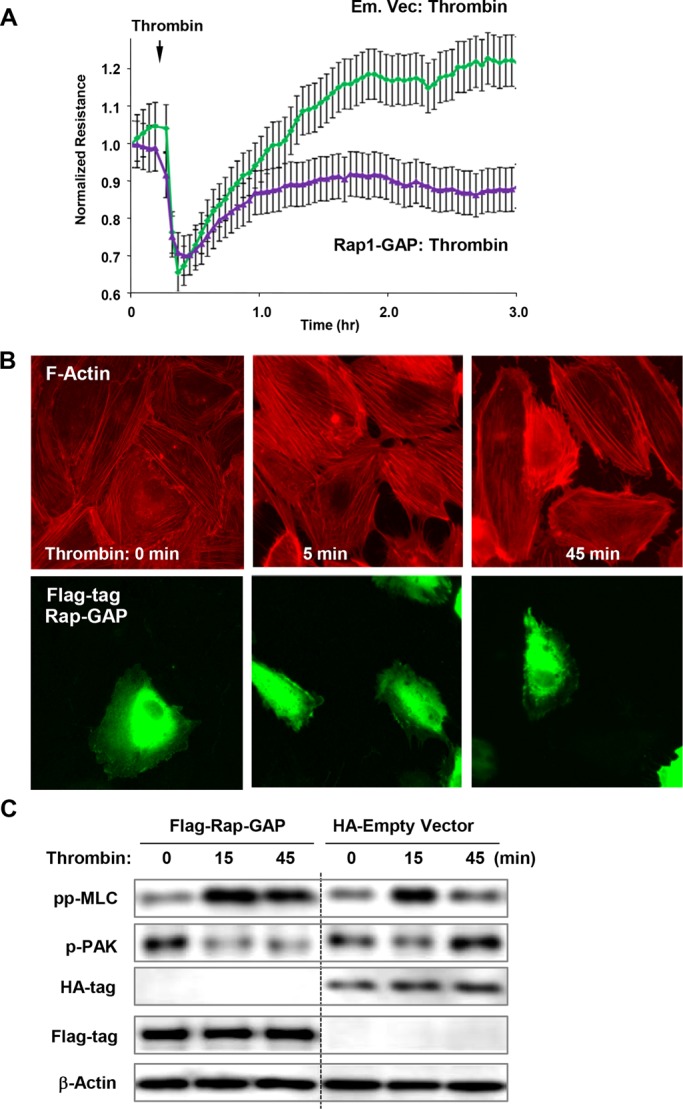
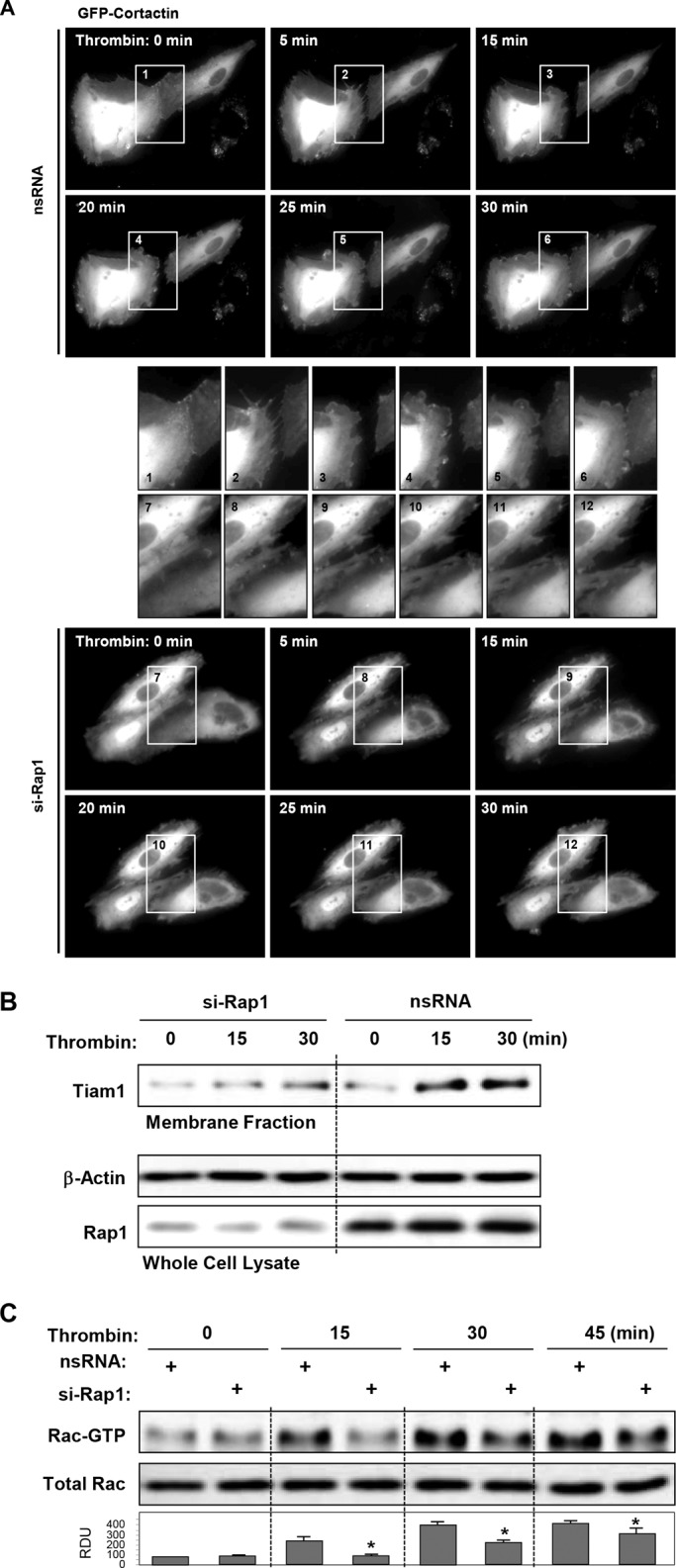
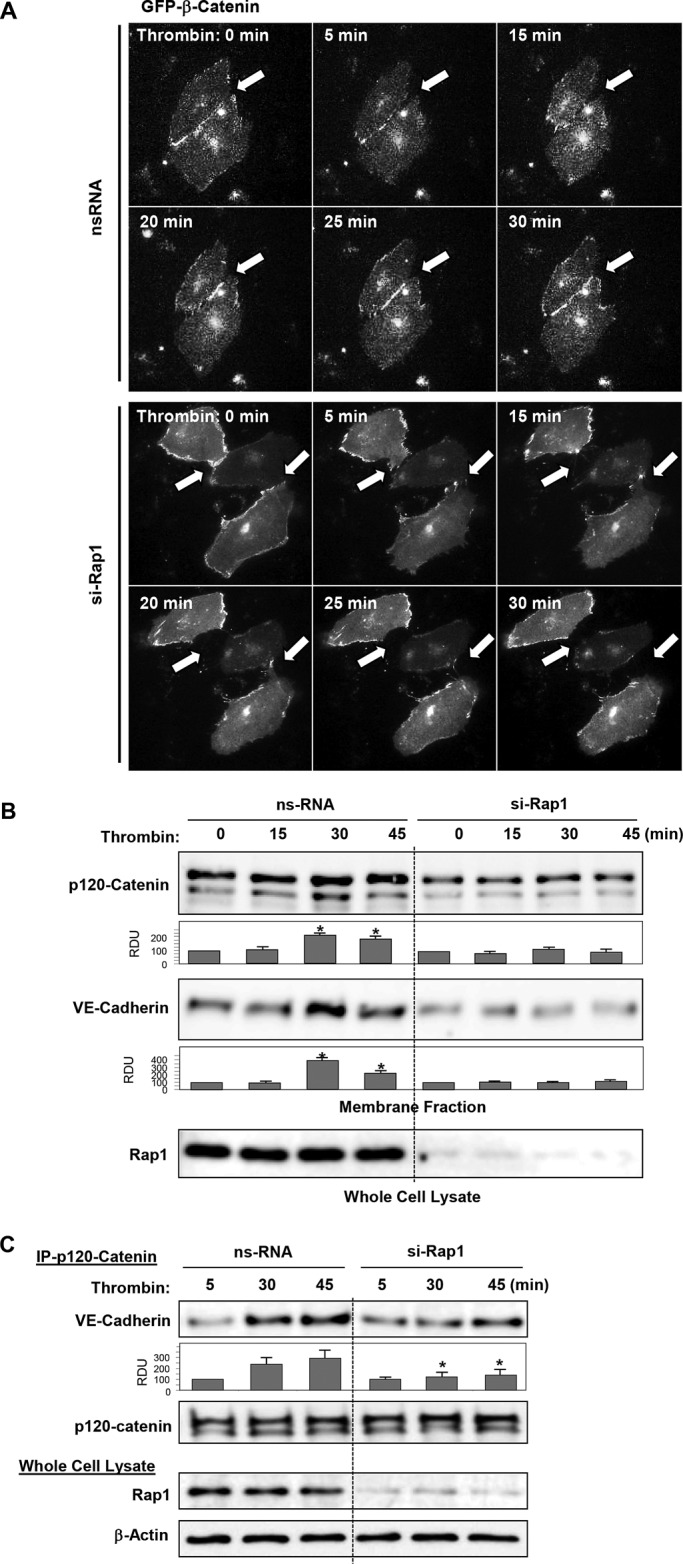
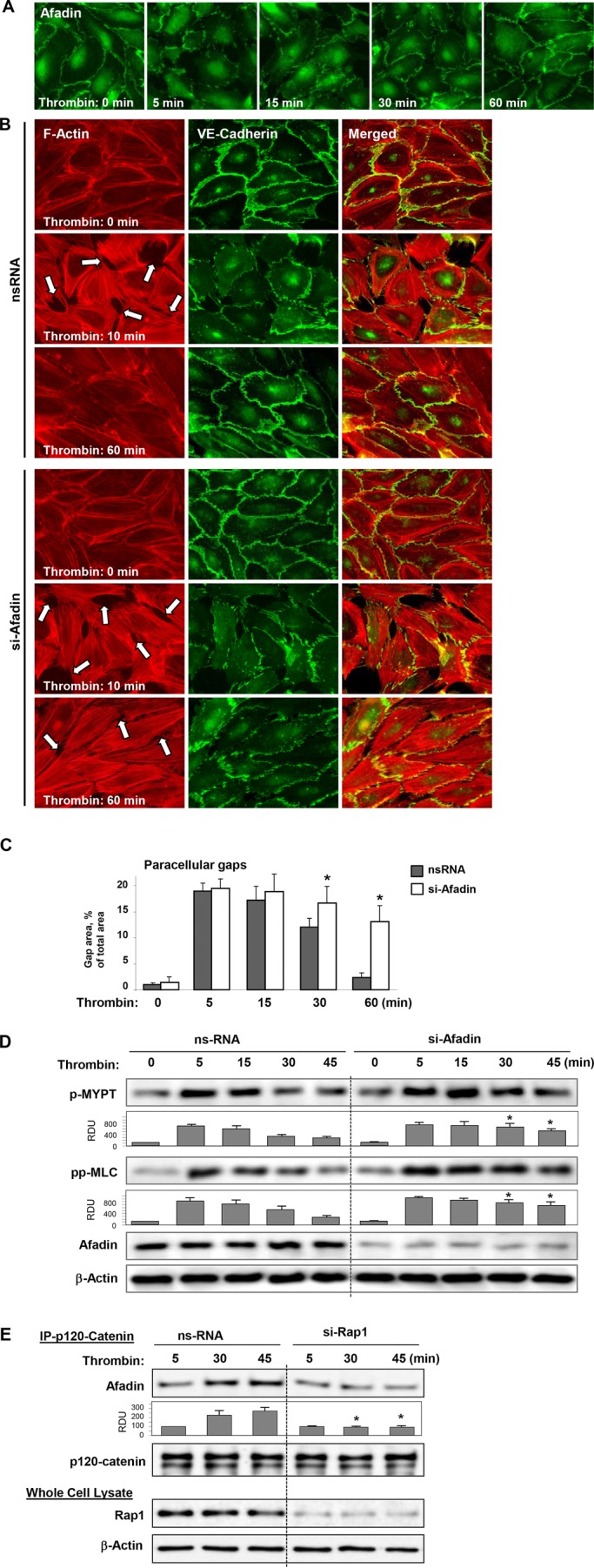
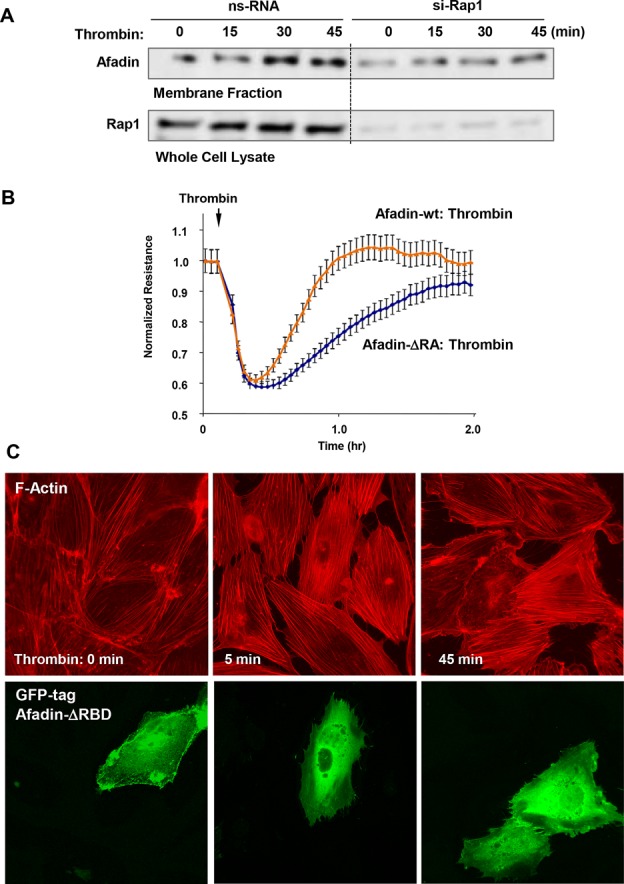
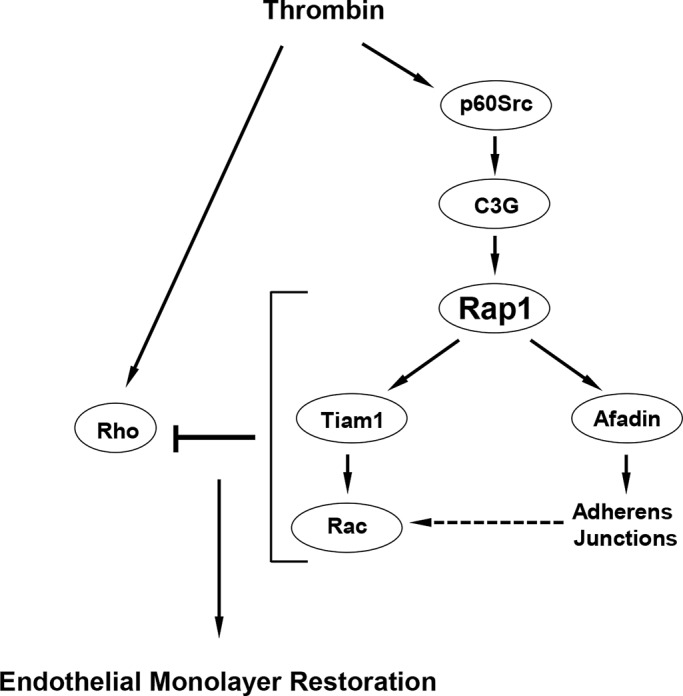
Activation of the Rho GTPase pathway determines endothelial cell (EC) hyperpermeability after injurious stimuli. To date, feedback mechanisms of Rho down-regulation critical for barrier restoration remain poorly understood. We tested a hypothesis that Rho down-regulation and barrier recovery of agonist-stimulated ECs is mediated by the Ras family GTPase Rap1. Thrombin-induced EC permeability driven by rapid activation of the Rho GTPase pathway was followed by Src kinase-dependent phosphorylation of the Rap1-specific guanine nucleotide exchange factor (GEF) C3G, activation of Rap1, and initiation of EC barrier recovery. Knockdown experiments showed that Rap1 activation was essential for down-regulation of Rho signaling and actin stress fiber dissolution. Rap1 activation also enhanced interaction between adherens junction (AJ) proteins VE-cadherin and p120-catenin and stimulated AJ reannealing mediated by the Rap1 effector afadin. This mechanism also included Rap1-dependent membrane translocation of the Rac1-specific GEF Tiam1 and activation of Rac1-dependent peripheral cytoskeletal dynamics, leading to resealing of intercellular gaps. These data demonstrate that activation of the Rap1-afadin axis is a physiological mechanism driving restoration of barrier integrity in agonist-stimulated EC monolayers via negative-feedback regulation of Rho signaling, stimulation of actin peripheral dynamics, and reestablishment of cell-cell adhesive complexes.
Figures
References
-
- Balzac F, Avolio M, Degani S, Kaverina I, Torti M, Silengo L, Small JV, Retta SF. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J Cell Sci. 2005;118:4765–4783. - PubMed
-
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
