

Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional fanconi anemia pathway
- PMID: 23867477
- PMCID: PMC3766423
- DOI: 10.1158/0008-5472.CAN-12-2975
Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional fanconi anemia pathway
Erratum in
-
Editor's Note: Werner Syndrome Helicase Has a Critical Role in DNA Damage Responses in the Absence of a Functional Fanconi Anemia Pathway.Cancer Res. 2025 Oct 1;85(19):3813. doi: 10.1158/0008-5472.CAN-25-3491. Cancer Res. 2025. PMID: 41030019 No abstract available.
Abstract
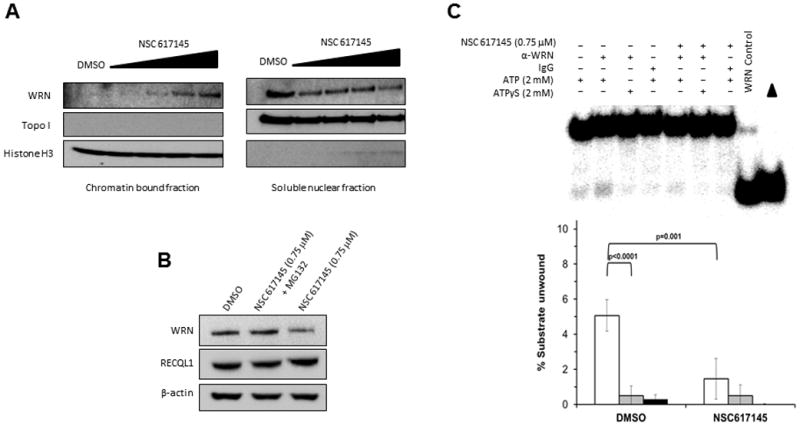
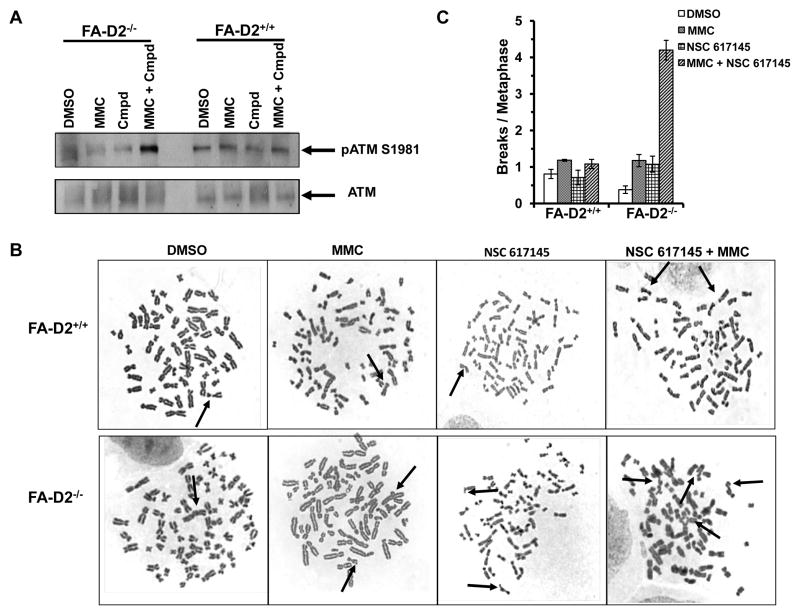
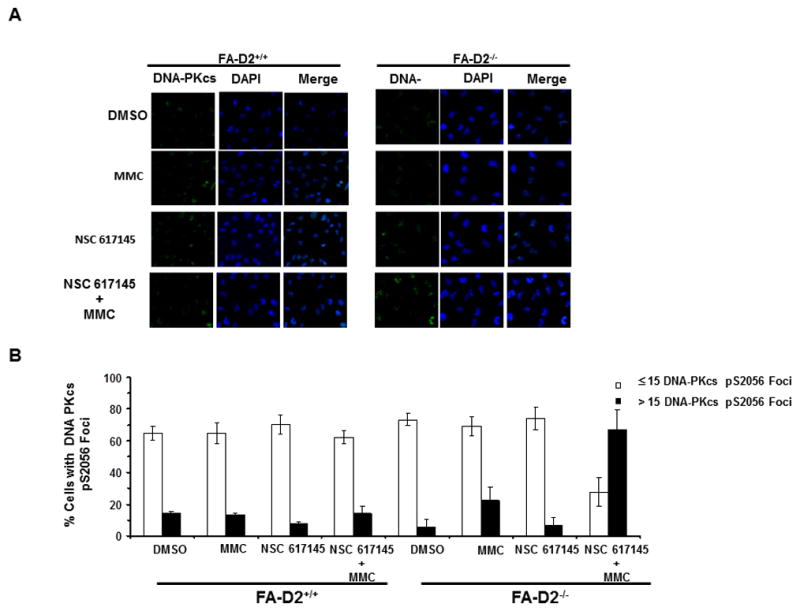
Werner syndrome is genetically linked to mutations in WRN that encodes a DNA helicase-nuclease believed to operate at stalled replication forks. Using a newly identified small-molecule inhibitor of WRN helicase (NSC 617145), we investigated the role of WRN in the interstrand cross-link (ICL) response in cells derived from patients with Fanconi anemia, a hereditary disorder characterized by bone marrow failure and cancer. In FA-D2(-/-) cells, NSC 617145 acted synergistically with very low concentrations of mitomycin C to inhibit proliferation in a WRN-dependent manner and induce double-strand breaks (DSB) and chromosomal abnormalities. Under these conditions, ataxia-telangiectasia mutated activation and accumulation of DNA-dependent protein kinase, catalytic subunit pS2056 foci suggested an increased number of DSBs processed by nonhomologous end-joining (NHEJ). Rad51 foci were also elevated in FA-D2(-/-) cells exposed to NSC 617145 and mitomycin C, suggesting that WRN helicase inhibition interferes with later steps of homologous recombination at ICL-induced DSBs. Thus, when the Fanconi anemia pathway is defective, WRN helicase inhibition perturbs the normal ICL response, leading to NHEJ activation. Potential implication for treatment of Fanconi anemia-deficient tumors by their sensitization to DNA cross-linking agents is discussed.
©2013 AACR.
Conflict of interest statement
No potential conflicts of interest.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
