

FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension
- PMID: 23867624
- PMCID: PMC3726153
- DOI: 10.1172/JCI65592
FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension
Abstract
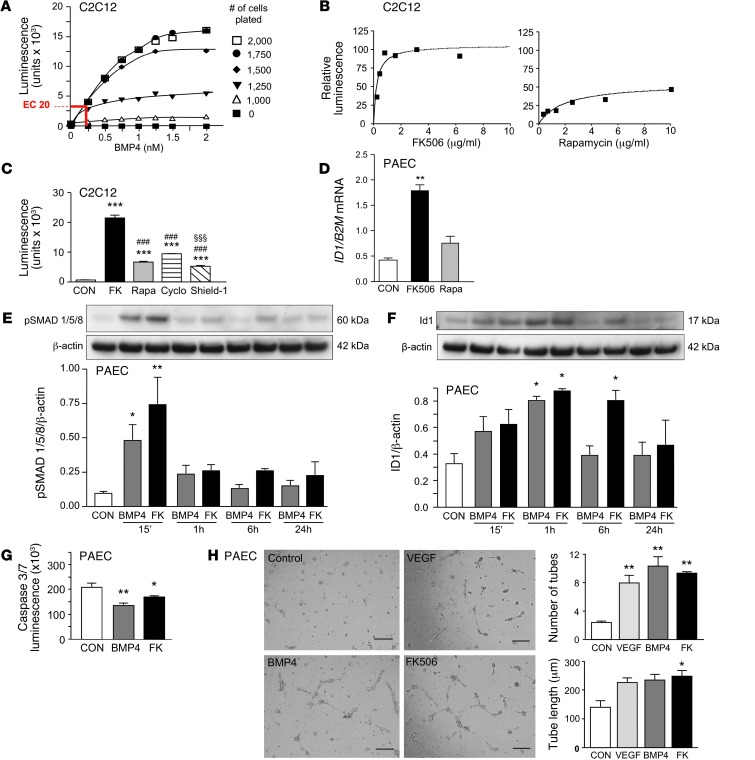
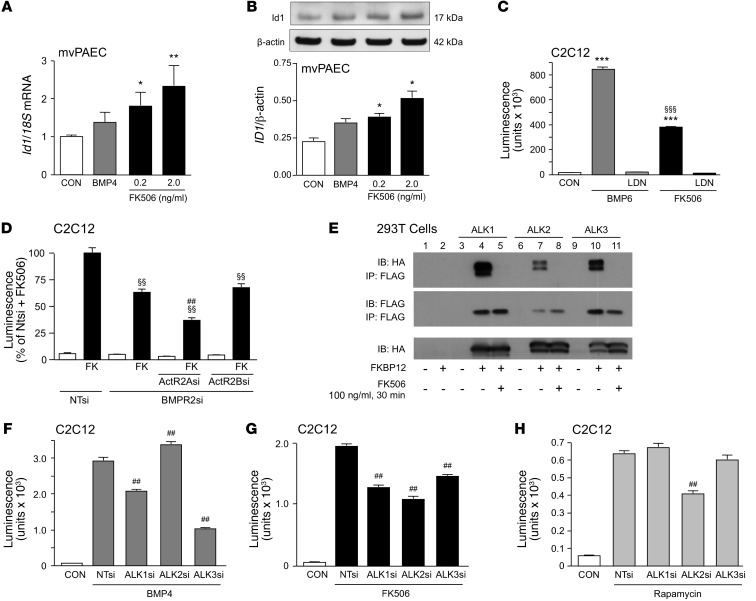
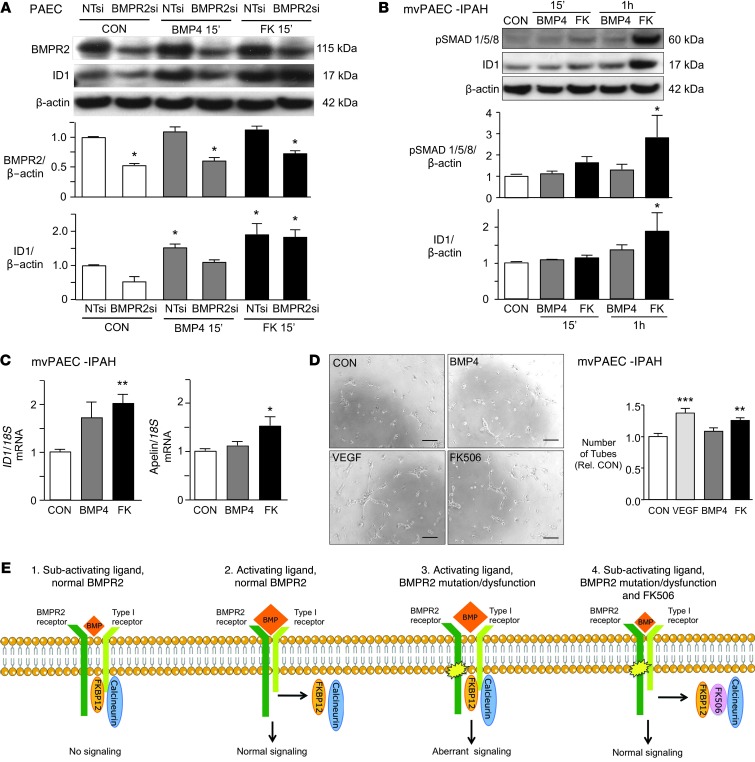
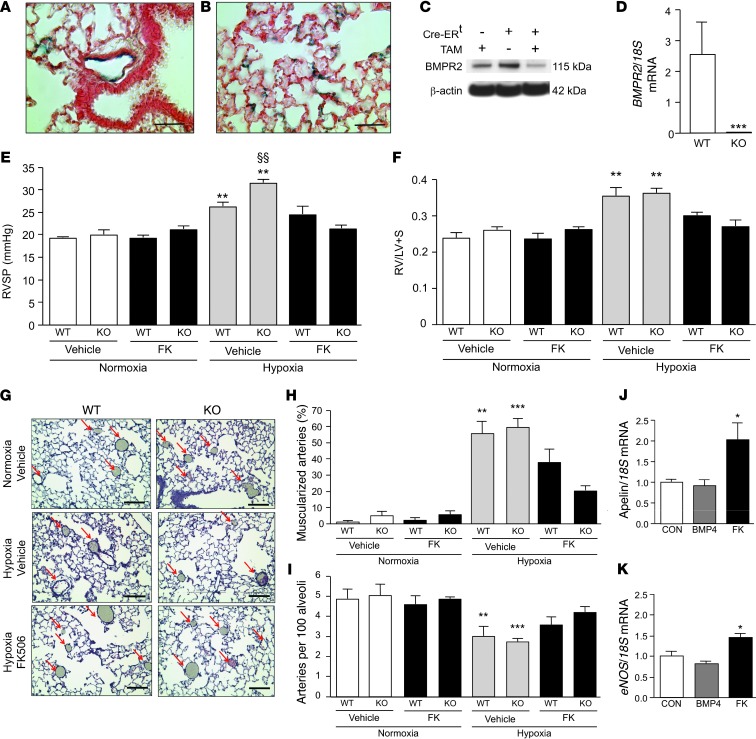
Dysfunctional bone morphogenetic protein receptor-2 (BMPR2) signaling is implicated in the pathogenesis of pulmonary arterial hypertension (PAH). We used a transcriptional high-throughput luciferase reporter assay to screen 3,756 FDA-approved drugs and bioactive compounds for induction of BMPR2 signaling. The best response was achieved with FK506 (tacrolimus), via a dual mechanism of action as a calcineurin inhibitor that also binds FK-binding protein-12 (FKBP12), a repressor of BMP signaling. FK506 released FKBP12 from type I receptors activin receptor-like kinase 1 (ALK1), ALK2, and ALK3 and activated downstream SMAD1/5 and MAPK signaling and ID1 gene regulation in a manner superior to the calcineurin inhibitor cyclosporine and the FKBP12 ligand rapamycin. In pulmonary artery endothelial cells (ECs) from patients with idiopathic PAH, low-dose FK506 reversed dysfunctional BMPR2 signaling. In mice with conditional Bmpr2 deletion in ECs, low-dose FK506 prevented exaggerated chronic hypoxic PAH associated with induction of EC targets of BMP signaling, such as apelin. Low-dose FK506 also reversed severe PAH in rats with medial hypertrophy following monocrotaline and in rats with neointima formation following VEGF receptor blockade and chronic hypoxia. Our studies indicate that low-dose FK506 could be useful in the treatment of PAH.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
