

Cardiac oxidative stress in a mouse model of neutral lipid storage disease
- PMID: 23867907
- PMCID: PMC3795454
- DOI: 10.1016/j.bbalip.2013.07.004
Cardiac oxidative stress in a mouse model of neutral lipid storage disease
Abstract
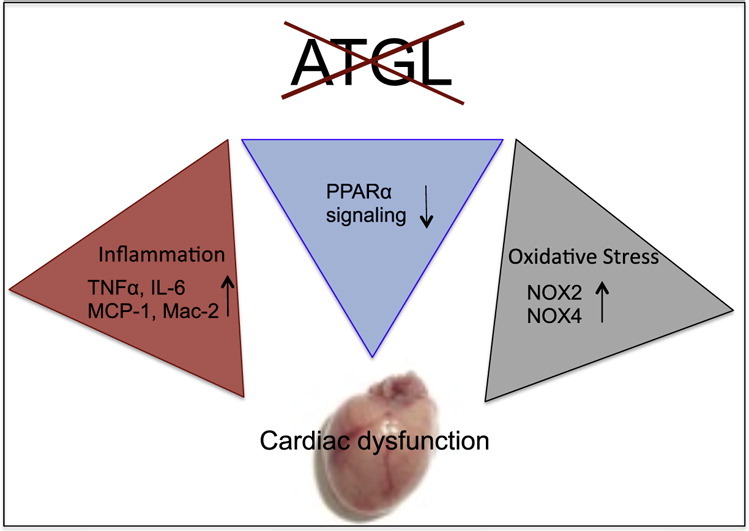
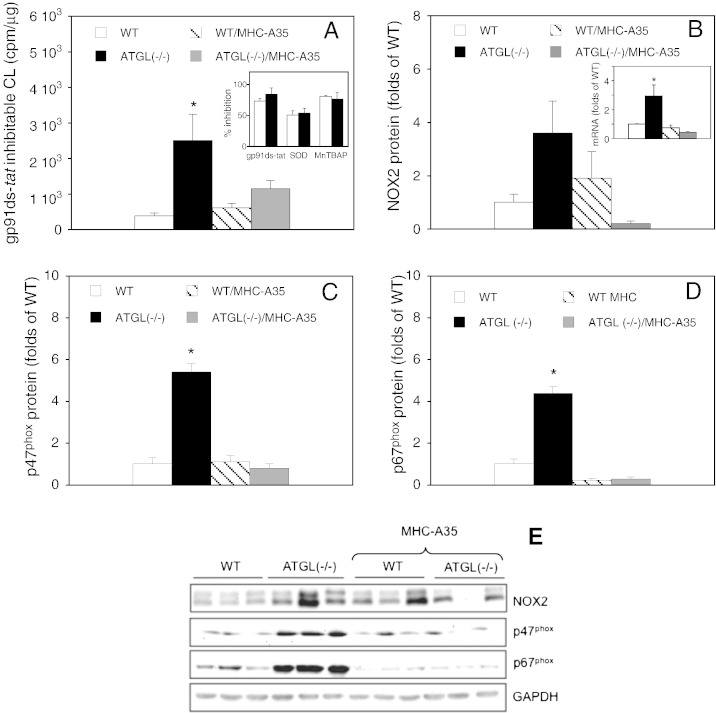
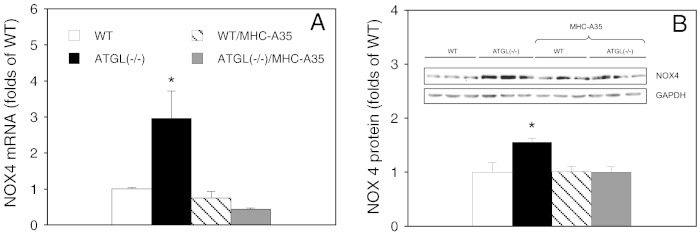
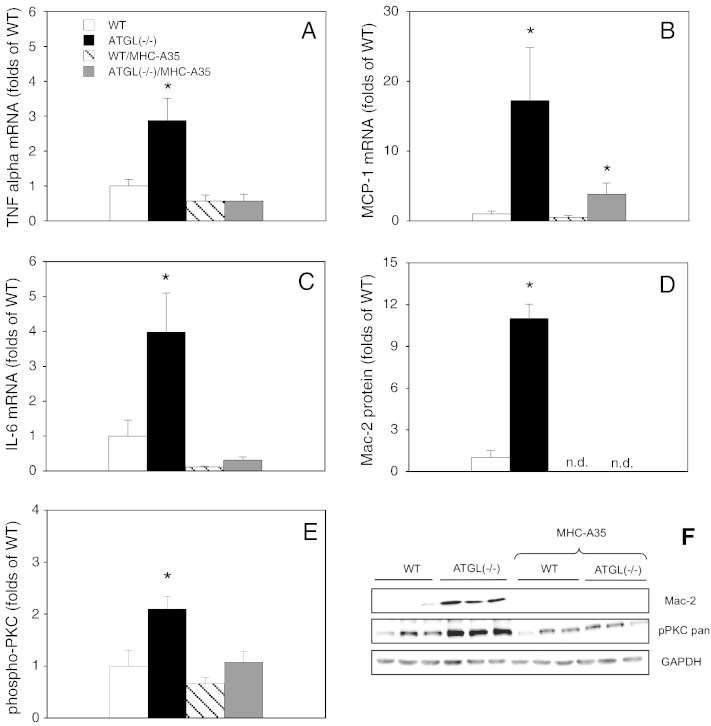
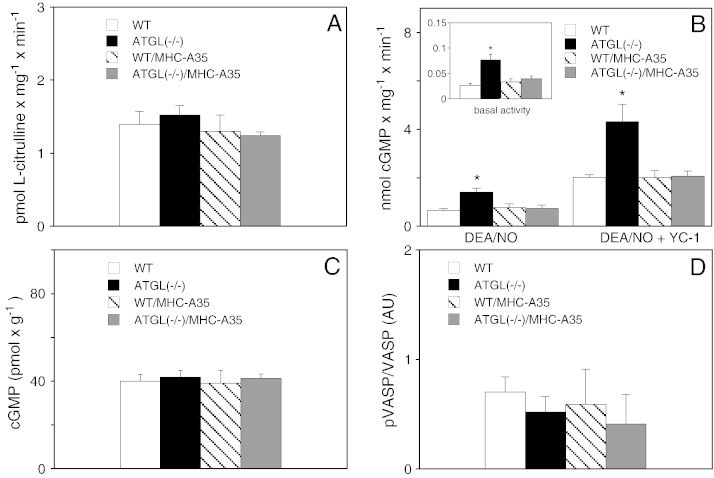
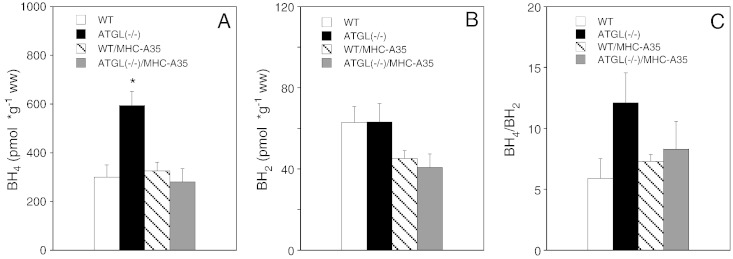
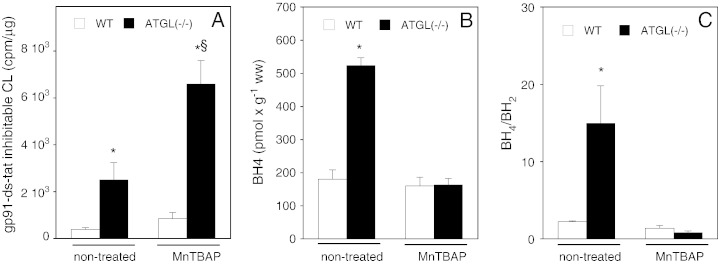
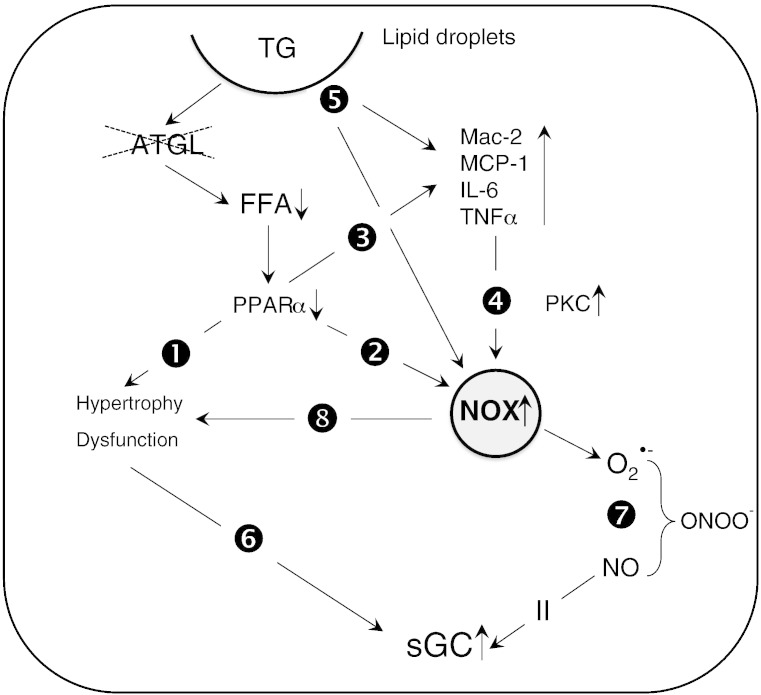
Cardiac oxidative stress has been implicated in the pathogenesis of hypertrophy, cardiomyopathy and heart failure. Systemic deletion of the gene encoding adipose triglyceride lipase (ATGL), the enzyme that catalyzes the rate-limiting step of triglyceride lipolysis, results in a phenotype characterized by severe steatotic cardiac dysfunction. The objective of the present study was to investigate a potential role of oxidative stress in cardiac ATGL deficiency. Hearts of mice with global ATGL knockout were compared to those of mice with cardiomyocyte-restricted overexpression of ATGL and to those of wildtype littermates. Our results demonstrate that oxidative stress, measured as lucigenin chemiluminescence, was increased ~6-fold in ATGL-deficient hearts. In parallel, cytosolic NADPH oxidase subunits p67phox and p47phox were upregulated 4-5-fold at the protein level. Moreover, a prominent upregulation of different inflammatory markers (tumor necrosis factor α, monocyte chemotactant protein-1, interleukin 6, and galectin-3) was observed in those hearts. Both the oxidative and inflammatory responses were abolished upon cardiomyocyte-restricted overexpression of ATGL. Investigating the effect of oxidative and inflammatory stress on nitric oxide/cGMP signal transduction we observed a ~2.5-fold upregulation of soluble guanylate cyclase activity and a ~2-fold increase in cardiac tetrahydrobiopterin levels. Systemic treatment of ATGL-deficient mice with the superoxide dismutase mimetic Mn(III)tetrakis (4-benzoic acid) porphyrin did not ameliorate but rather aggravated cardiac oxidative stress. Our data suggest that oxidative and inflammatory stress seems involved in lipotoxic heart disease. Upregulation of soluble guanylate cyclase and cardiac tetrahydrobiopterin might be regarded as counterregulatory mechanisms in cardiac ATGL deficiency.
Keywords: (s)GC; (soluble) guanylate cyclase; 2,2-diethyl-1-nitroso-oxyhydrazine; ATGL; ATGL(−/−); Adipose triglyceride lipase; BH(2); BH(4); Cardiac hypertrophy; DAG; DEA/NO; FFA; GAPDH; IL-6; Inflammation; MCP-1; Mac-2; Mn(III)tetrakis (4-benzoic acid) porphyrin chloride; MnTBAP; NADPH; NADPH oxidase; NO; NOX; ONOO(−); Oxidative stress; PBS; PKC; PPARα; SOD; TG; TNFα; VASP; adipose triglyceride lipase; adipose triglyceride lipase knockout; diacylglycerol; dihydrobiopterin, [2-amino-6-(1,2-dihydroxypropyl)-7,8-dihydro-1H-pteridin-4-one]; eNOS; endothelial nitric oxide synthase; free fatty acid; galectin-3; glyceraldehyde-3-phosphate dehydrogenase; iNOS; inducible nitric oxide synthase; interleukin 6; monocyte chemotactic protein-1; nNOS; neuronal nitric oxide synthase; nicotinamide adenine dinucleotide phosphate; nitric oxide; pVASP; peroxisome proliferator receptor α; peroxynitrite; phosphate-buffered saline; phosphorylated vasodilator-stimulated phosphoprotein; protein kinase C; superoxide dismutase; tetrahydrobiopterin, [(6R)-2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydropteridin-4(1H)-one]; triacylglycerol; tumor necrosis factor α; vasodilator-stimulated phosphoprotein.
© 2013. Published by Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Role of the ubiquitin-proteasome system in cardiac dysfunction of adipose triglyceride lipase-deficient mice.J Mol Cell Cardiol. 2014 Dec;77:11-9. doi: 10.1016/j.yjmcc.2014.09.028. Epub 2014 Oct 5. J Mol Cell Cardiol. 2014. PMID: 25285770 Free PMC article.
-
Endothelial dysfunction in adipose triglyceride lipase deficiency.Biochim Biophys Acta. 2014 Jun;1841(6):906-17. doi: 10.1016/j.bbalip.2014.03.005. Epub 2014 Mar 21. Biochim Biophys Acta. 2014. PMID: 24657704 Free PMC article.
-
Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy.Diabetes. 2013 May;62(5):1464-77. doi: 10.2337/db12-0927. Epub 2013 Jan 24. Diabetes. 2013. PMID: 23349479 Free PMC article.
-
The role of adipose triglyceride lipase in lipid and glucose homeostasis: lessons from transgenic mice.Lipids Health Dis. 2019 Nov 22;18(1):204. doi: 10.1186/s12944-019-1151-z. Lipids Health Dis. 2019. PMID: 31757217 Free PMC article. Review.
-
Critical roles for α/β hydrolase domain 5 (ABHD5)/comparative gene identification-58 (CGI-58) at the lipid droplet interface and beyond.Biochim Biophys Acta Mol Cell Biol Lipids. 2017 Oct;1862(10 Pt B):1233-1241. doi: 10.1016/j.bbalip.2017.07.016. Epub 2017 Aug 4. Biochim Biophys Acta Mol Cell Biol Lipids. 2017. PMID: 28827091 Free PMC article. Review.
Cited by
-
Mn porphyrin-based SOD mimic, MnTnHex-2-PyP(5+), and non-SOD mimic, MnTBAP(3-), suppressed rat spinal cord ischemia/reperfusion injury via NF-κB pathways.Free Radic Res. 2014 Dec;48(12):1426-42. doi: 10.3109/10715762.2014.960865. Epub 2014 Oct 10. Free Radic Res. 2014. PMID: 25185063 Free PMC article.
-
The nutrigenetic influence of the interaction between dietary vitamin E and TXN and COMT gene polymorphisms on waist circumference: a case control study.J Transl Med. 2015 Sep 2;13:286. doi: 10.1186/s12967-015-0652-4. J Transl Med. 2015. PMID: 26329592 Free PMC article.
-
Adipose triglyceride lipase decrement affects skeletal muscle homeostasis during aging through FAs-PPARα-PGC-1α antioxidant response.Oncotarget. 2016 Apr 26;7(17):23019-32. doi: 10.18632/oncotarget.8552. Oncotarget. 2016. PMID: 27056902 Free PMC article.
-
ATGL deficiency aggravates pressure overload-triggered myocardial hypertrophic remodeling associated with the proteasome-PTEN-mTOR-autophagy pathway.Cell Biol Toxicol. 2023 Oct;39(5):2113-2131. doi: 10.1007/s10565-022-09699-0. Epub 2022 Feb 26. Cell Biol Toxicol. 2023. PMID: 35218467 Free PMC article.
-
Outside-in signaling by femoral cuff injury induces a distinct vascular lesion in adipose triglyceride lipase knockout mice.Histol Histopathol. 2021 Jan;36(1):91-100. doi: 10.14670/HH-18-285. Epub 2020 Nov 24. Histol Histopathol. 2021. PMID: 33231284
References
-
- Murdoch C.E., Zhang M., Cave A.C., Shah A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006;71:208–215. - PubMed
-
- Takimoto E., Champion H.C., Li M., Ren S., Rodriguez E.R., Tavazzi B., Lazzarino G., Paolocci N., Gabrielson K.L., Wang Y., Kass D.A. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Invest. 2005;115:1221–1231. - PMC - PubMed
-
- Engberding N., Spiekermann S., Schaefer A., Heineke A., Wiencke A., Muller M., Fuchs M., Hilfiker-Kleiner D., Hornig B., Drexler H., Landmesser U. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation. 2004;110:2175–2179. - PubMed
-
- Feillet-Coudray C., Sutra T., Fouret G., Ramos J., Wrutniak-Cabello C., Cabello G., Cristol J.P., Coudray C. Oxidative stress in rats fed a high-fat high-sucrose diet and preventive effect of polyphenols: involvement of mitochondrial and NAD(P)H oxidase systems. Free Radic. Biol. Med. 2009;46:624–632. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
