

The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling
- PMID: 23870130
- PMCID: PMC3753670
- DOI: 10.1016/j.cell.2013.06.016
The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling
Abstract
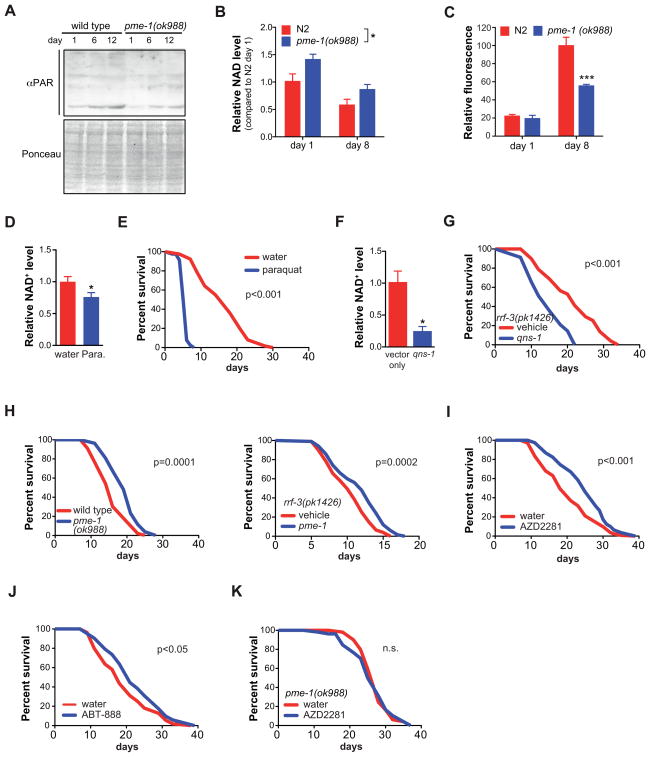
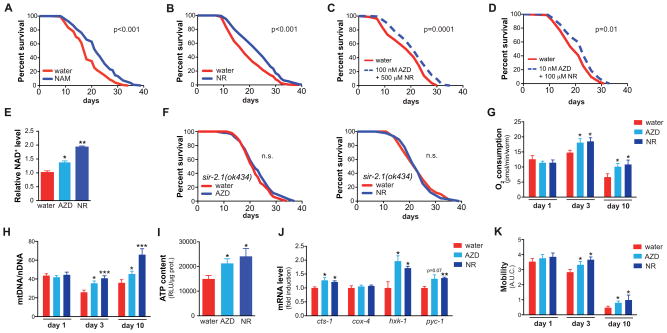
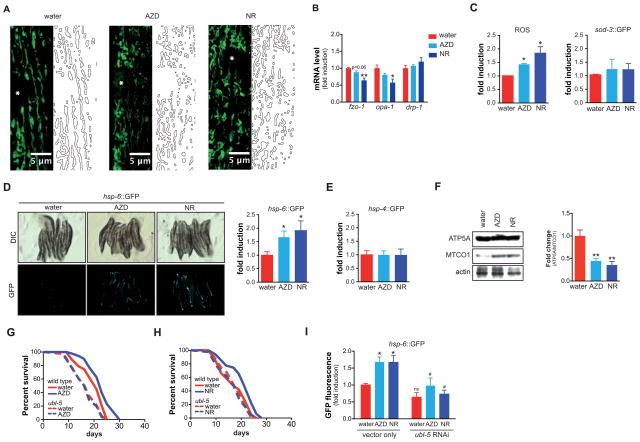
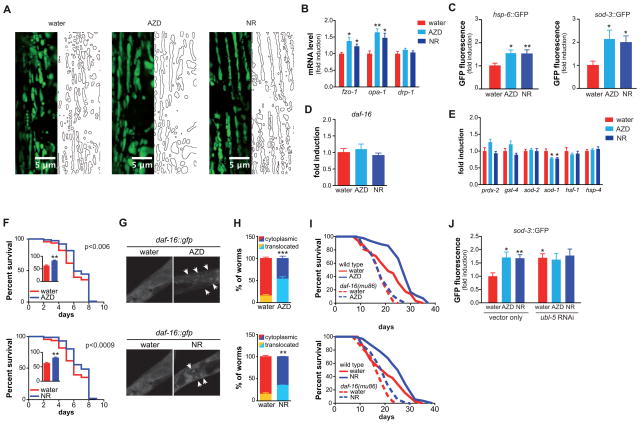
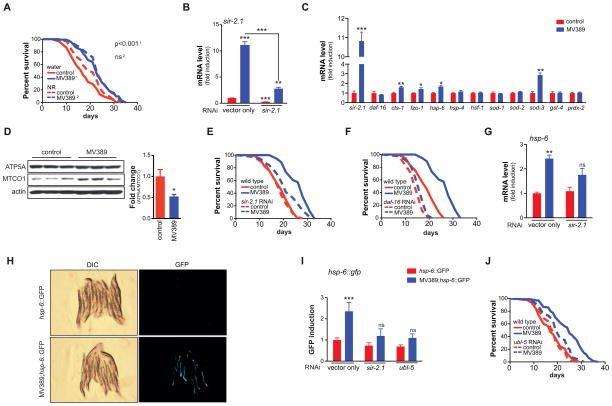
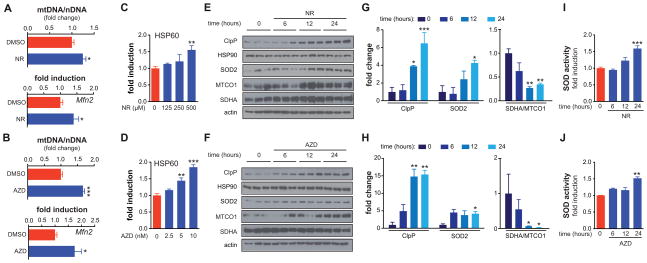
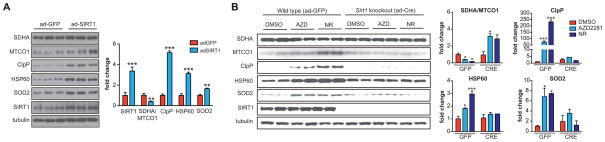
NAD(+) is an important cofactor regulating metabolic homeostasis and a rate-limiting substrate for sirtuin deacylases. We show that NAD(+) levels are reduced in aged mice and Caenorhabditis elegans and that decreasing NAD(+) levels results in a further reduction in worm lifespan. Conversely, genetic or pharmacological restoration of NAD(+) prevents age-associated metabolic decline and promotes longevity in worms. These effects are dependent upon the protein deacetylase sir-2.1 and involve the induction of mitonuclear protein imbalance as well as activation of stress signaling via the mitochondrial unfolded protein response (UPR(mt)) and the nuclear translocation and activation of FOXO transcription factor DAF-16. Our data suggest that augmenting mitochondrial stress signaling through the modulation of NAD(+) levels may be a target to improve mitochondrial function and prevent or treat age-associated decline.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Aging: seeking mitonuclear balance.Cell. 2013 Jul 18;154(2):271-3. doi: 10.1016/j.cell.2013.06.046. Cell. 2013. PMID: 23870118
-
Sirtuins: Longevity focuses on NAD+.Nat Chem Biol. 2013 Nov;9(11):666-7. doi: 10.1038/nchembio.1369. Nat Chem Biol. 2013. PMID: 24141218 No abstract available.
References
-
- Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–953. - PubMed
-
- Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P, Chini EN. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. Faseb J. 2007;21:3629–3639. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
