

The footprint of genome architecture in the largest genome expansion in RNA viruses
- PMID: 23874204
- PMCID: PMC3715407
- DOI: 10.1371/journal.ppat.1003500
The footprint of genome architecture in the largest genome expansion in RNA viruses
Abstract
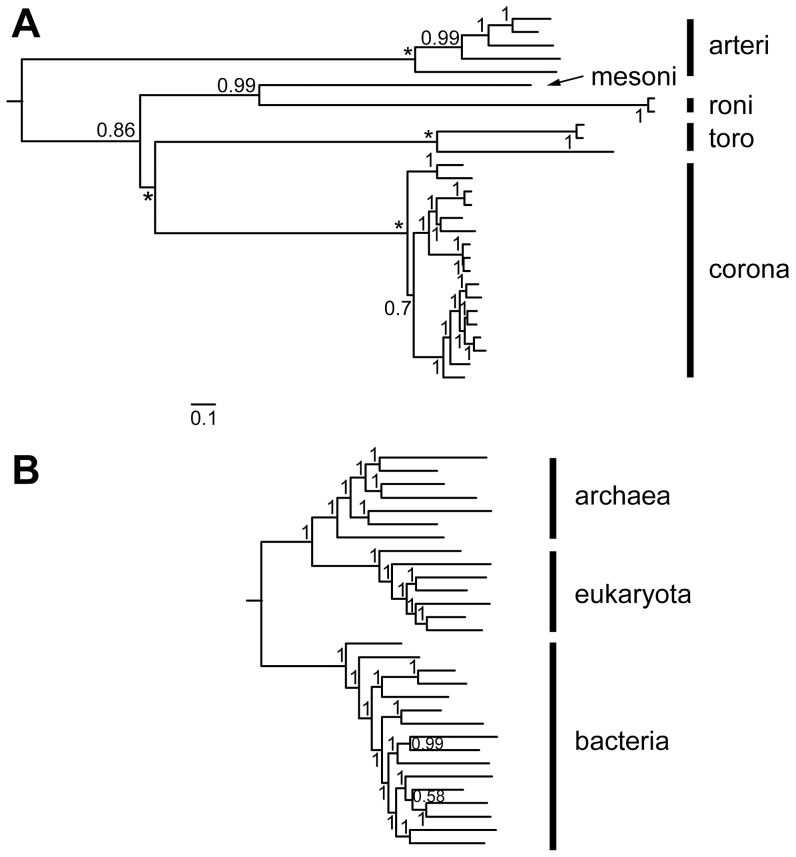
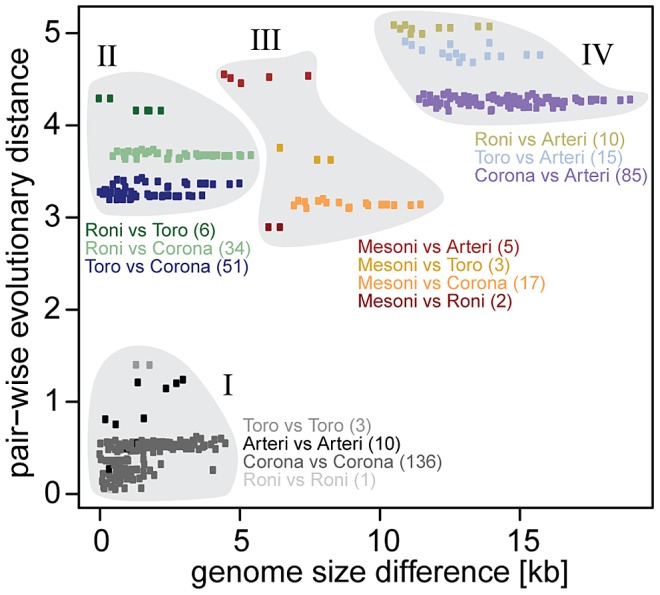
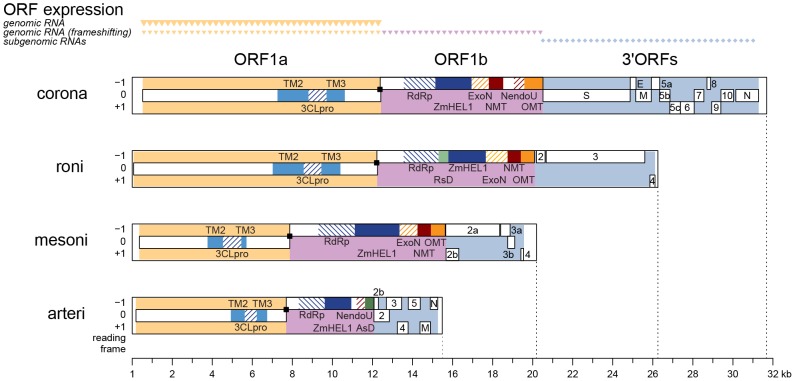
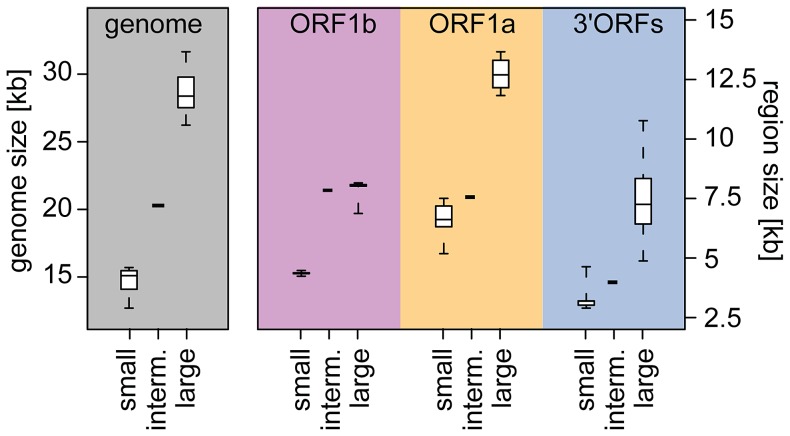
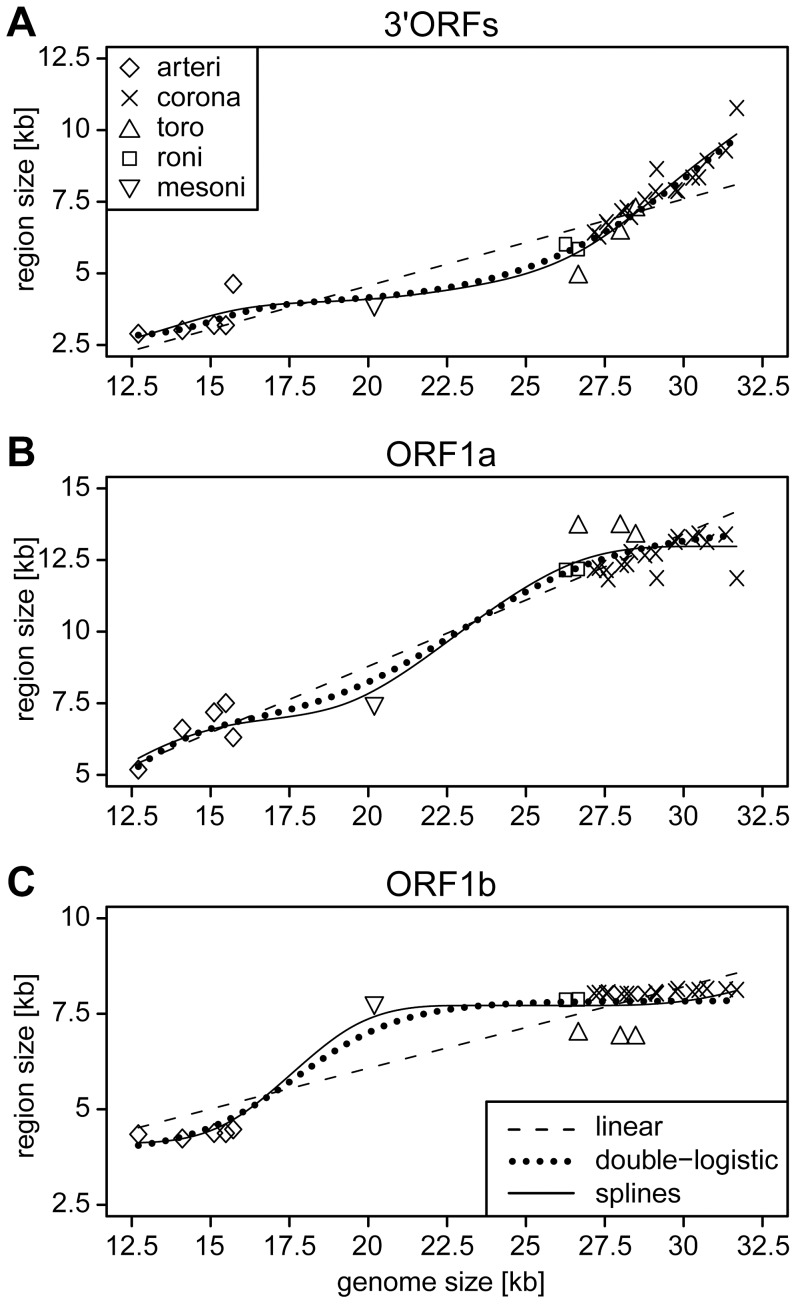
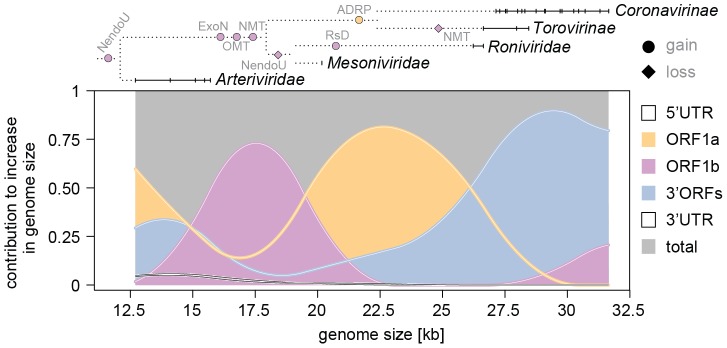
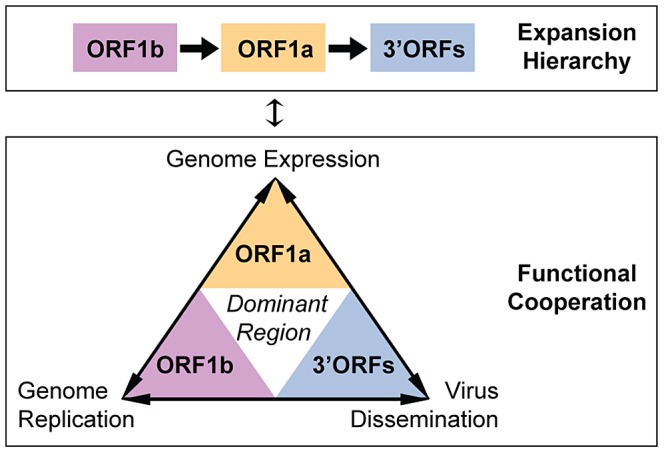
The small size of RNA virus genomes (2-to-32 kb) has been attributed to high mutation rates during replication, which is thought to lack proof-reading. This paradigm is being revisited owing to the discovery of a 3'-to-5' exoribonuclease (ExoN) in nidoviruses, a monophyletic group of positive-stranded RNA viruses with a conserved genome architecture. ExoN, a homolog of canonical DNA proof-reading enzymes, is exclusively encoded by nidoviruses with genomes larger than 20 kb. All other known non-segmented RNA viruses have smaller genomes. Here we use evolutionary analyses to show that the two- to three-fold expansion of the nidovirus genome was accompanied by a large number of replacements in conserved proteins at a scale comparable to that in the Tree of Life. To unravel common evolutionary patterns in such genetically diverse viruses, we established the relation between genomic regions in nidoviruses in a sequence alignment-free manner. We exploited the conservation of the genome architecture to partition each genome into five non-overlapping regions: 5' untranslated region (UTR), open reading frame (ORF) 1a, ORF1b, 3'ORFs (encompassing the 3'-proximal ORFs), and 3' UTR. Each region was analyzed for its contribution to genome size change under different models. The non-linear model statistically outperformed the linear one and captured >92% of data variation. Accordingly, nidovirus genomes were concluded to have reached different points on an expansion trajectory dominated by consecutive increases of ORF1b, ORF1a, and 3'ORFs. Our findings indicate a unidirectional hierarchical relation between these genome regions, which are distinguished by their expression mechanism. In contrast, these regions cooperate bi-directionally on a functional level in the virus life cycle, in which they predominantly control genome replication, genome expression, and virus dissemination, respectively. Collectively, our findings suggest that genome architecture and the associated region-specific division of labor leave a footprint on genome expansion and may limit RNA genome size.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Szathmary E, Smith JM (1995) The Major Evolutionary Transitions. Nature 374: 227–232. - PubMed
-
- Lynch M, Conery JS (2003) The origins of genome complexity. Science 302: 1401–1404. - PubMed
-
- Sniegowski PD, Gerrish PJ, Johnson T, Shaver A (2000) The evolution of mutation rates: separating causes from consequences. Bioessays 22: 1057–1066. - PubMed
-
- Gago S, Elena SF, Flores R, Sanjuan R (2009) Extremely High Mutation Rate of a Hammerhead Viroid. Science 323: 1308. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
