

SMETANA: accurate and scalable algorithm for probabilistic alignment of large-scale biological networks
- PMID: 23874484
- PMCID: PMC3710069
- DOI: 10.1371/journal.pone.0067995
SMETANA: accurate and scalable algorithm for probabilistic alignment of large-scale biological networks
Abstract
In this paper we introduce an efficient algorithm for alignment of multiple large-scale biological networks. In this scheme, we first compute a probabilistic similarity measure between nodes that belong to different networks using a semi-Markov random walk model. The estimated probabilities are further enhanced by incorporating the local and the cross-species network similarity information through the use of two different types of probabilistic consistency transformations. The transformed alignment probabilities are used to predict the alignment of multiple networks based on a greedy approach. We demonstrate that the proposed algorithm, called SMETANA, outperforms many state-of-the-art network alignment techniques, in terms of computational efficiency, alignment accuracy, and scalability. Our experiments show that SMETANA can easily align tens of genome-scale networks with thousands of nodes on a personal computer without any difficulty. The source code of SMETANA is available upon request. The source code of SMETANA can be downloaded from http://www.ece.tamu.edu/~bjyoon/SMETANA/.
Conflict of interest statement
Figures
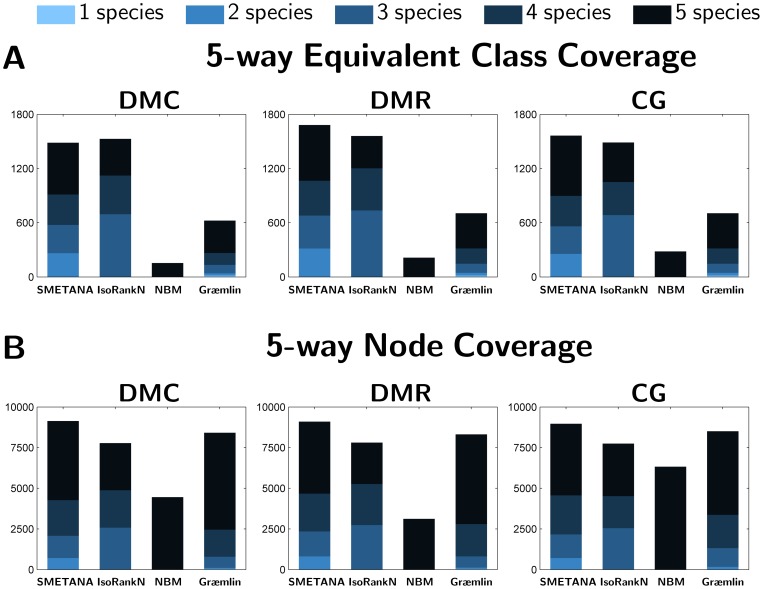
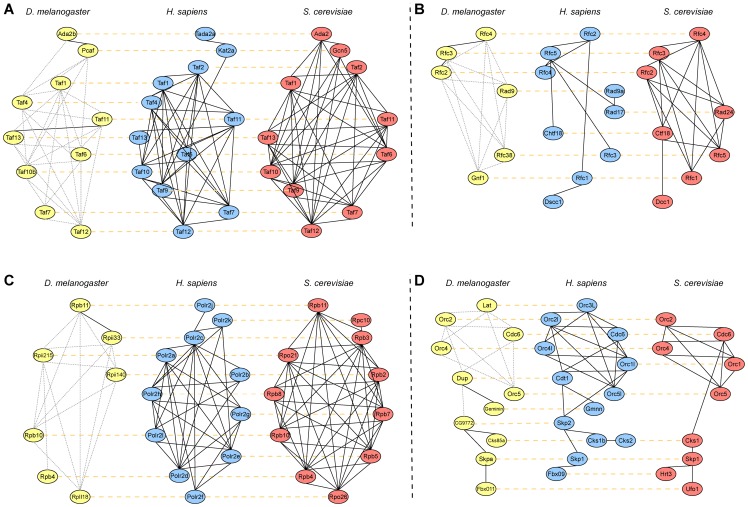
 species (
species ( ). (B) Node Coverage: Number of nodes (i.e. proteins) that belong to equivalence classes that contain nodes from
). (B) Node Coverage: Number of nodes (i.e. proteins) that belong to equivalence classes that contain nodes from  species in the 5-way alignment.
species in the 5-way alignment.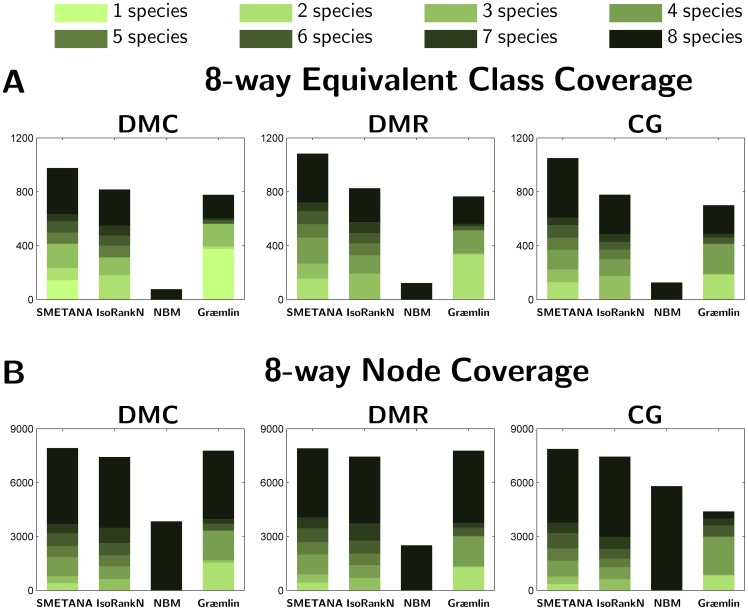
 species (
species ( ). (B) Node Coverage: Number of nodes (i.e. proteins) that belong to equivalence classes that contain nodes from
). (B) Node Coverage: Number of nodes (i.e. proteins) that belong to equivalence classes that contain nodes from  species in the 8-way alignment.
species in the 8-way alignment.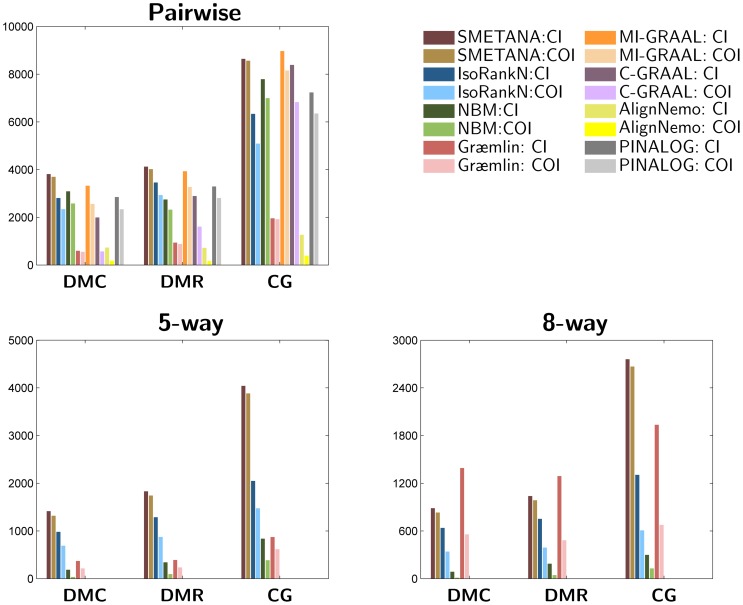

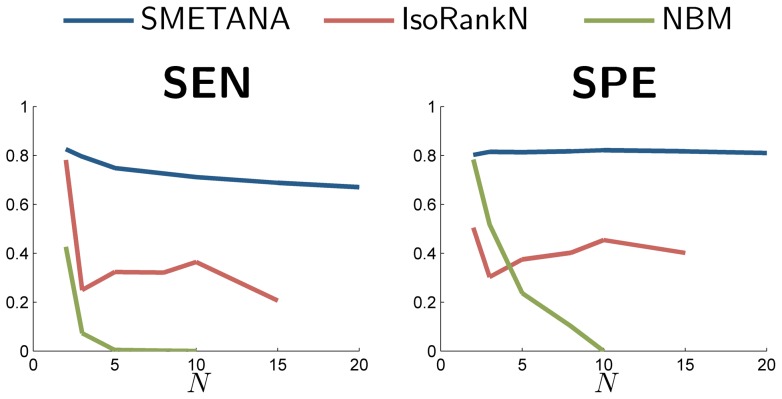
 increases the separation between the two score distributions, which increases the discriminative power of the node similarity score for predicting potential orthologs. Measures reported: specificity (SPE), number of correct nodes (CN) (which reflects the sensitivity), mean normalized entropy (MNE), number of conserved interactions (CI), number of conserved orthologous interactions (COI).
increases the separation between the two score distributions, which increases the discriminative power of the node similarity score for predicting potential orthologs. Measures reported: specificity (SPE), number of correct nodes (CN) (which reflects the sensitivity), mean normalized entropy (MNE), number of conserved interactions (CI), number of conserved orthologous interactions (COI).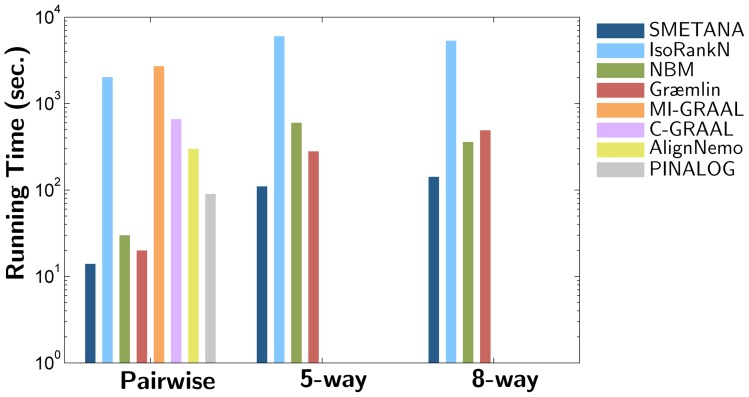
References
-
- Zhang A (2009) Protein Interaction Networks: Computational Analysis. New York, NY, USA: Cambridge University Press, 1st edition.
-
- Barabasi AL, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113. - PubMed
-
- Cusick ME, Klitgord N, Vidal M, Hill DE (2005) Interactome: gateway into systems biology. Hum Mol Genet 14 Spec No. 2: R171–181. - PubMed
-
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627. - PubMed
-
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, et al. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
