

New metrics for comparison of taxonomies reveal striking discrepancies among species delimitation methods in Madascincus lizards
- PMID: 23874561
- PMCID: PMC3710018
- DOI: 10.1371/journal.pone.0068242
New metrics for comparison of taxonomies reveal striking discrepancies among species delimitation methods in Madascincus lizards
Abstract
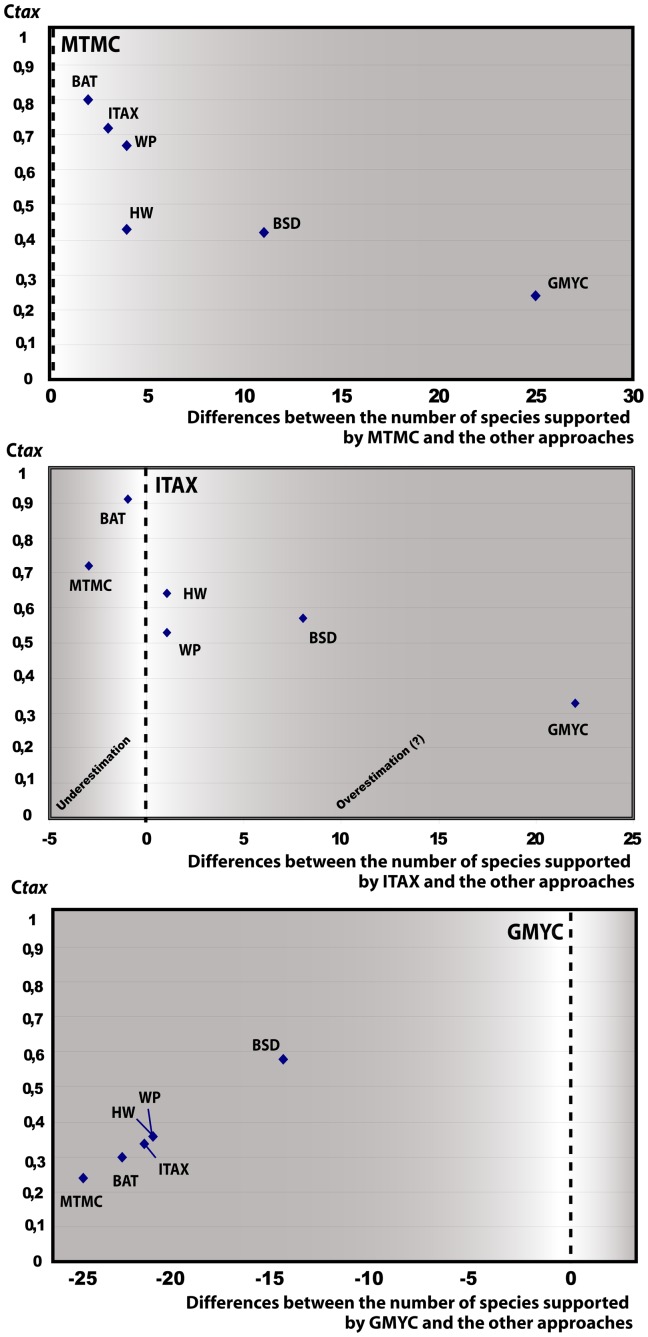
Delimiting and describing species is fundamental to numerous biological disciplines such as evolution, macroecology, and conservation. Delimiting species as independent evolutionary lineages may and often does yield different outcomes depending on the species criteria applied, but methods should be chosen that minimize the inference of objectively erroneous species limits. Several protocols exploit single-gene or multi-gene coalescence statistics, assignment tests or other rationales related to nuclear DNA (nDNA) allele sharing to automatically delimit species. We apply seven different species delimitation protocols to a taxonomically confusing group of Malagasy lizards (Madascincus), and compare the resulting taxonomies with two newly developed metrics: the Taxonomic index of congruence C tax which quantifies the congruence between two taxonomies, and the Relative taxonomic resolving power index R tax which quantifies the potential of an approach to capture a high number of species boundaries. The protocols differed in the total number of species proposed, between 9 and 34, and were also highly incongruent in placing species boundaries. The Generalized Mixed Yule-Coalescent approach captured the highest number of potential species boundaries but many of these were clearly contradicted by extensive nDNA admixture between sympatric mitochondrial DNA (mtDNA) haplotype lineages. Delimiting species as phenotypically diagnosable mtDNA clades failed to detect two cryptic species that are unambiguous due to a lack of nDNA gene flow despite sympatry. We also consider the high number of species boundaries and their placement by multi-gene Bayesian species delimitation as poorly reliable whereas the Bayesian assignment test approach provided a species delimitation highly congruent with integrative taxonomic practice. The present study illustrates the trade-off in taxonomy between reliability (favored by conservative approaches) and resolving power (favored by inflationist approaches). Quantifying excessive splitting is more difficult than quantifying excessive lumping, suggesting a priority for conservative taxonomies in which errors are more liable to be detected and corrected by subsequent studies.
Conflict of interest statement
Figures






References
-
- Wheeler QD, Knapp S, Stevenson DW, Stevenson J, Blum SD, et al. (2012) Mapping the biosphere: Exploring species to understand the origin, organization, and sustainability of biodiversity. Syst Biodivers 10: 1–20.
-
- Areekul Butcher B, Quicke DLJ (2012) A turbo-taxonomic study of Thai Aleiodes (Aleiodes) and Aleiodes (Arcaleiodes) (Hymenoptera: Braconidae: Rogadinae) based largely on COI barcoded specimens, with rapid descriptions of 179 new species. Zootaxa 3457: 1–232.
-
- Costello MJ, May RM, Stork NE (2013) Can we name Earth’s species before they go extinct? Science 339: 413–416. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
