

Selective constraint, background selection, and mutation accumulation variability within and between human populations
- PMID: 23875710
- PMCID: PMC3727949
- DOI: 10.1186/1471-2164-14-495
Selective constraint, background selection, and mutation accumulation variability within and between human populations
Abstract
Background: Regions of the genome that are under evolutionary constraint across multiple species have previously been used to identify functional sequences in the human genome. Furthermore, it is known that there is an inverse relationship between evolutionary constraint and the allele frequency of a mutation segregating in human populations, implying a direct relationship between interspecies divergence and fitness in humans. Here we utilise this relationship to test differences in the accumulation of putatively deleterious mutations both between populations and on the individual level.
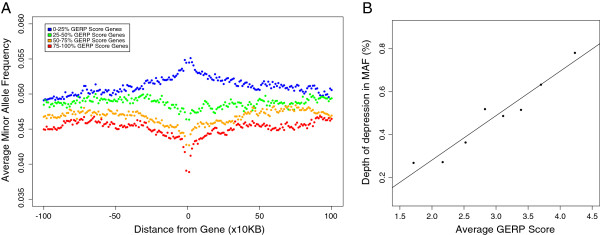
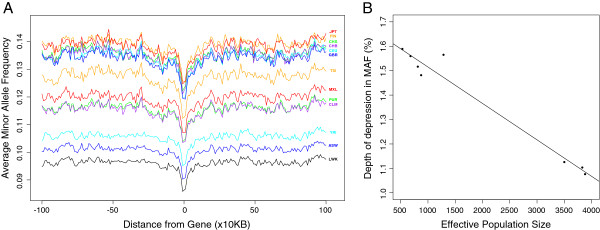
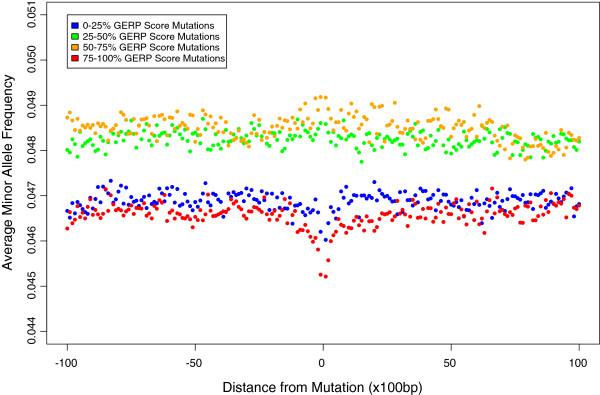
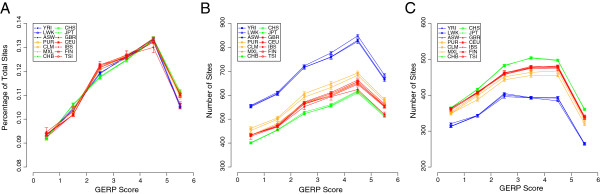
Results: Using whole genome and exome sequencing data from Phase 1 of the 1000 Genome Project for 1,092 individuals from 14 worldwide populations we show that minor allele frequency (MAF) varies as a function of constraint around both coding regions and non-coding sites genome-wide, implying that negative, rather than positive, selection primarily drives the distribution of alleles among individuals via background selection. We find a strong relationship between effective population size and the depth of depression in MAF around the most conserved genes, suggesting that populations with smaller effective size are carrying more deleterious mutations, which also translates into higher genetic load when considering the number of putatively deleterious alleles segregating within each population. Finally, given the extreme richness of the data, we are now able to classify individual genomes by the accumulation of mutations at functional sites using high coverage 1000 Genomes data. Using this approach we detect differences between 'healthy' individuals within populations for the distributions of putatively deleterious rare alleles they are carrying.
Conclusions: These findings demonstrate the extent of background selection in the human genome and highlight the role of population history in shaping patterns of diversity between human individuals. Furthermore, we provide a framework for the utility of personal genomic data for the study of genetic fitness and diseases.
Figures
Similar articles
-
Recombination Rate Variation, Hitchhiking, and Demographic History Shape Deleterious Load in Poplar.Mol Biol Evol. 2016 Nov;33(11):2899-2910. doi: 10.1093/molbev/msw169. Epub 2016 Aug 10. Mol Biol Evol. 2016. PMID: 27512114
-
Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome.PLoS Genet. 2011 Oct;7(10):e1002326. doi: 10.1371/journal.pgen.1002326. Epub 2011 Oct 13. PLoS Genet. 2011. PMID: 22022285 Free PMC article.
-
Purifying selection in deeply conserved human enhancers is more consistent than in coding sequences.PLoS One. 2014 Jul 25;9(7):e103357. doi: 10.1371/journal.pone.0103357. eCollection 2014. PLoS One. 2014. PMID: 25062004 Free PMC article.
-
Rates and fitness consequences of new mutations in humans.Genetics. 2012 Feb;190(2):295-304. doi: 10.1534/genetics.111.134668. Genetics. 2012. PMID: 22345605 Free PMC article. Review.
-
Comparative genomics and the study of evolution by natural selection.Mol Ecol. 2008 Nov;17(21):4586-96. doi: 10.1111/j.1365-294X.2008.03954.x. Mol Ecol. 2008. PMID: 19140982 Review.
Cited by
-
Genetic Variation in Pan Species Is Shaped by Demographic History and Harbors Lineage-Specific Functions.Genome Biol Evol. 2019 Apr 1;11(4):1178-1191. doi: 10.1093/gbe/evz047. Genome Biol Evol. 2019. PMID: 30847478 Free PMC article.
-
Properties of human disease genes and the role of genes linked to Mendelian disorders in complex disease aetiology.Hum Mol Genet. 2017 Feb 1;26(3):489-500. doi: 10.1093/hmg/ddw405. Hum Mol Genet. 2017. PMID: 28053046 Free PMC article.
-
Understanding the Hidden Complexity of Latin American Population Isolates.Am J Hum Genet. 2018 Nov 1;103(5):707-726. doi: 10.1016/j.ajhg.2018.09.013. Epub 2018 Oct 25. Am J Hum Genet. 2018. PMID: 30401458 Free PMC article.
-
Whole-exome sequencing reveals a rapid change in the frequency of rare functional variants in a founding population of humans.PLoS Genet. 2013;9(9):e1003815. doi: 10.1371/journal.pgen.1003815. Epub 2013 Sep 26. PLoS Genet. 2013. PMID: 24086152 Free PMC article.
-
REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants.Am J Hum Genet. 2016 Oct 6;99(4):877-885. doi: 10.1016/j.ajhg.2016.08.016. Epub 2016 Sep 22. Am J Hum Genet. 2016. PMID: 27666373 Free PMC article.
References
-
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE. et al.Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. - DOI - PMC - PubMed
-
- Ep C. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636–640. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
