

Mucopolysaccharidosis IVA: correlation between genotype, phenotype and keratan sulfate levels
- PMID: 23876334
- PMCID: PMC3779837
- DOI: 10.1016/j.ymgme.2013.06.008
Mucopolysaccharidosis IVA: correlation between genotype, phenotype and keratan sulfate levels
Abstract
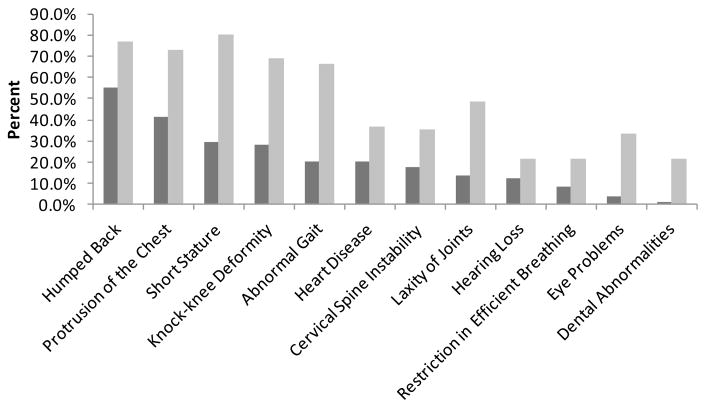
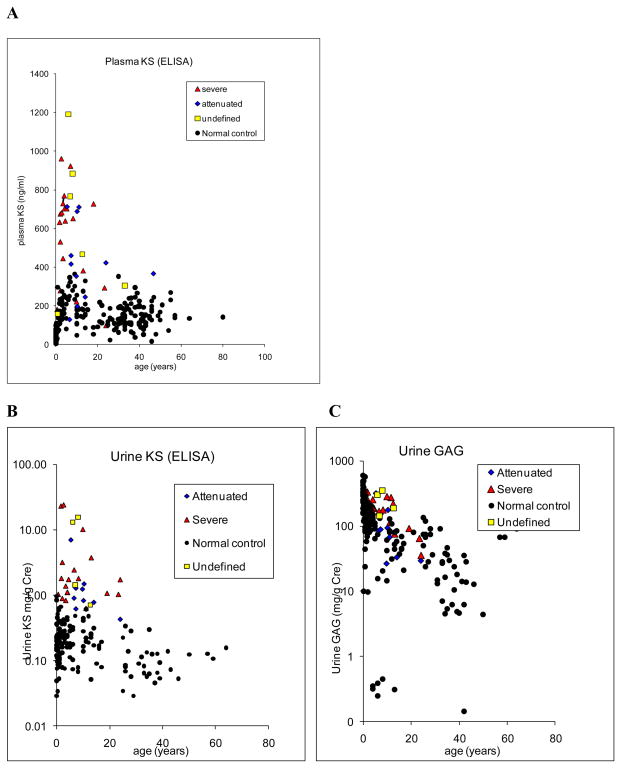
Mucopolysaccharidosis IVA (MPS IVA) is caused by deficiency of N-acetylgalactosamine-6-sulfate sulfatase (GALNS), leading to systemic skeletal dysplasia because of excessive storage of keratan sulfate (KS) in chondrocytes. In an effort to determine a precise prognosis and personalized treatment, we aim to characterize clinical, biochemical, and molecular findings in MPS IVA patients, and to seek correlations between genotype, phenotype, and blood and urine KS levels. Mutation screening of GALNS gene was performed in 55 MPS IVA patients (severe: 36, attenuated: 13, undefined: 6) by genomic PCR followed by direct sequence analysis. Plasma and urine KS levels were measured by ELISA method. Genotype/phenotype/KS correlations were assessed when data were available. Fifty-three different mutations including 19 novel ones (41 missense, 2 nonsense, 4 small deletions, 1 insertion, and 5 splice-site) were identified in 55 patients and accounted for 93.6% of the analyzed mutant alleles. Thirty-nine mutations were associated with a severe phenotype and ten mutations with an attenuated one. Blood and urine KS concentrations in MPS IVA patients were age-dependent and markedly higher than those in age-matched normal controls. Plasma and urine KS levels in MPS IVA patients with the severe phenotype were higher than in those with an attenuated form. This study provides evidence for extensive allelic heterogeneity of MPS IVA. Accumulation of mutations as well as clinical descriptions and KS levels allows us to predict clinical severity more precisely and should be used for evaluation of responses to potential treatment options.
Keywords: Biomarker; C6S; CDC; Centers for Disease Control and Prevention; Chondroitin-6-sulfate; Cr; Creatinine; DMB; DMSO; GAGs; GALNS; Genotype; Glycosaminoglycans; IMO; IVA Mucopolysaccharidosis IVA; International Morquio Organization; KS; Keratan sulfate; LSD; Lysosomal storage disorder; MPS; Mucopolysaccharidosis IVA; N-acetylgalactosamine-6-sulfate sufatase; Phenotype; dimethylmethylene blue; dimethylsulfoxide.
© 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Lowry RB, Applegarth DA, Toone JR, MacDonald E, Thunem NY. An update on the frequency of mucopolysaccharide syndromes in British Columbia. Hum Genet. 1990;85:389–390. - PubMed
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. - PubMed
-
- Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997;101:355–358. - PubMed
-
- Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003;123:310–313. - PubMed
-
- Pinto R, Caseiro C, Lemos M, Lopes L, Fontes A, Ribeiro H, Pinto E, Silva E, Rocha S, Marcao A, Ribeiro I, Lacerda L, Ribeiro G, Amaral O, Sa Miranda MC. Prevalence of lysosomal storage diseases in Portugal. Eur J Hum Genet. 2004;12:87–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
