

Viral degradasome hijacks mitochondria to suppress innate immunity
- PMID: 23877405
- PMCID: PMC3731571
- DOI: 10.1038/cr.2013.98
Viral degradasome hijacks mitochondria to suppress innate immunity
Abstract
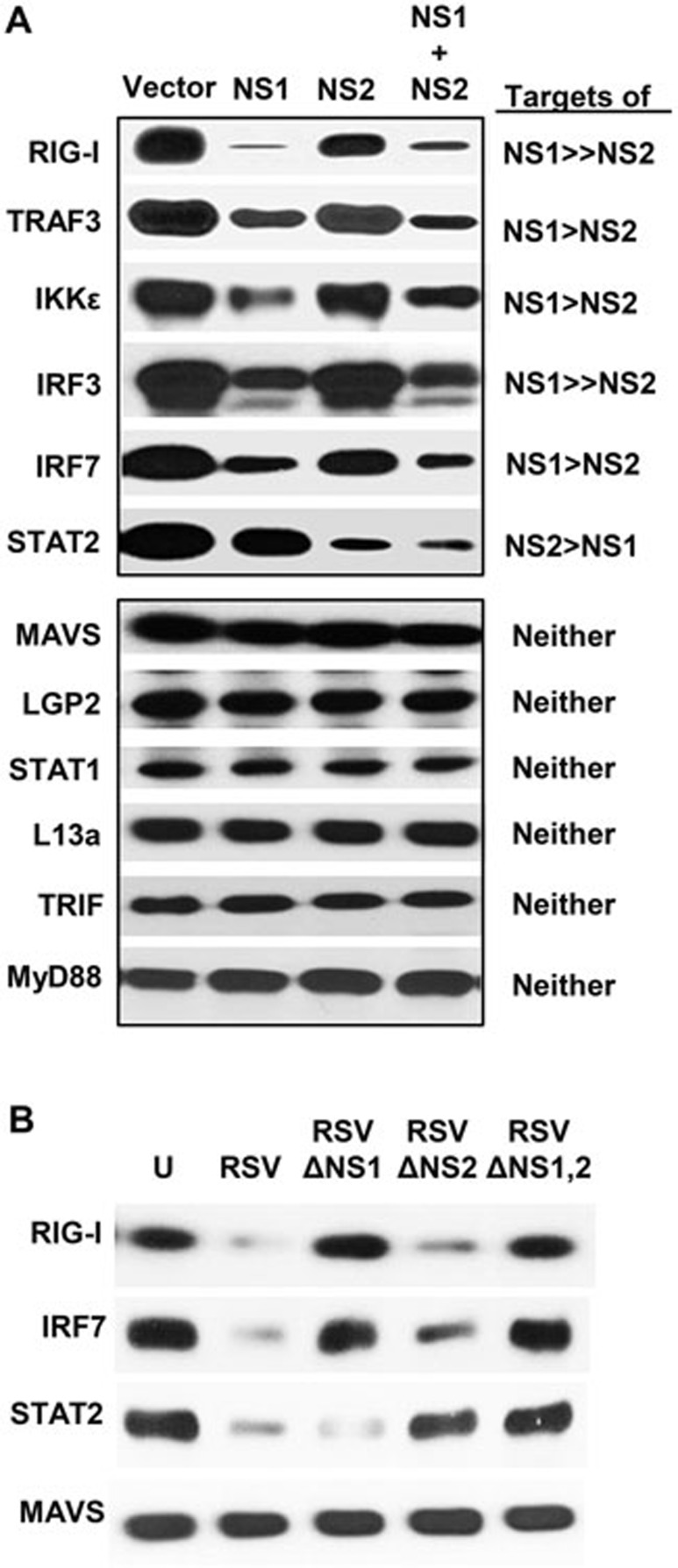
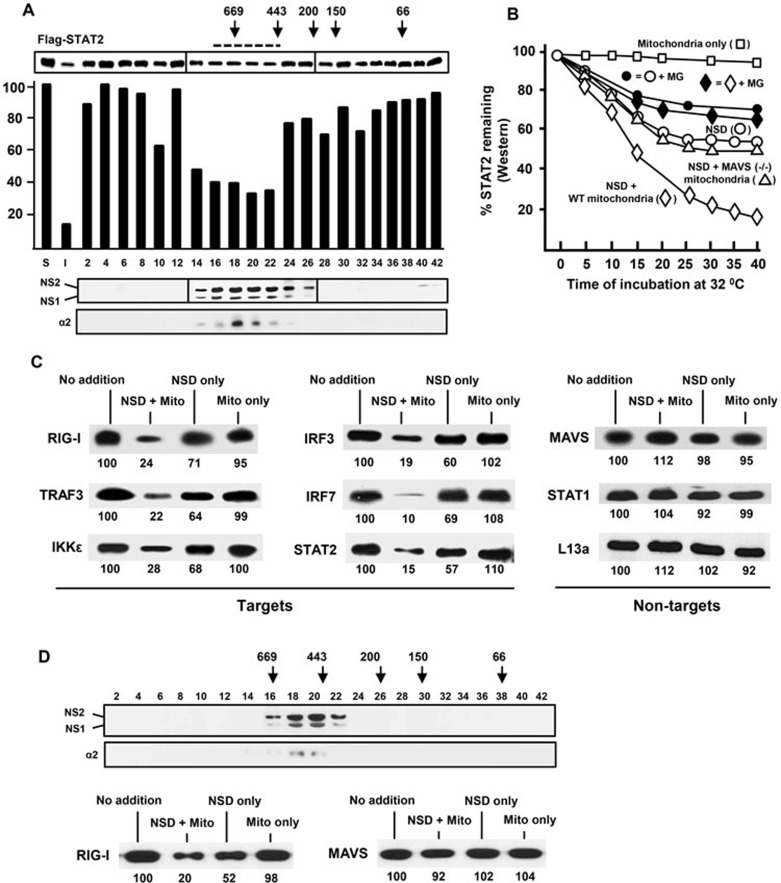
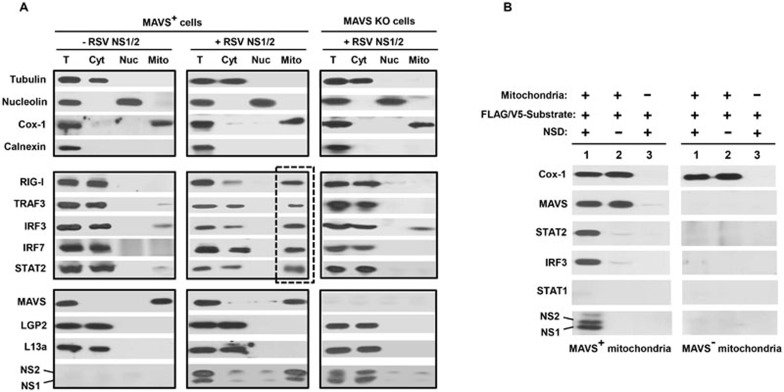
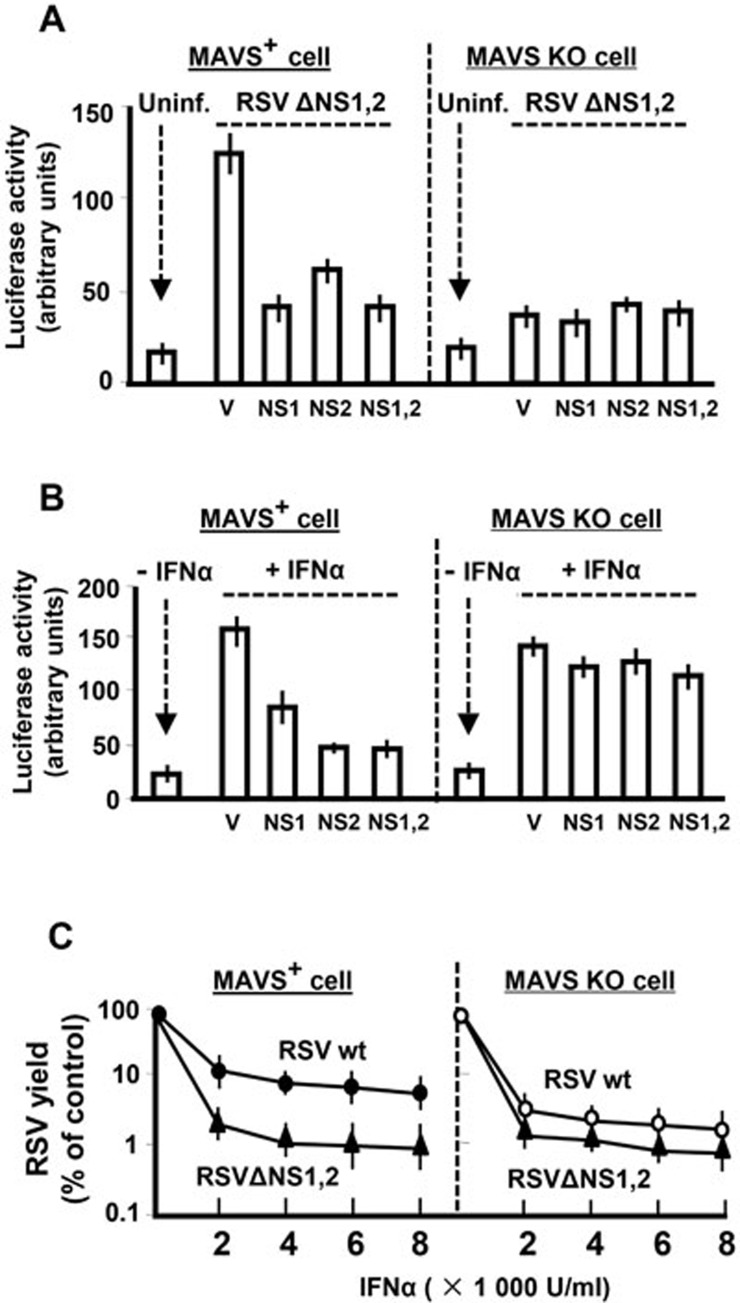
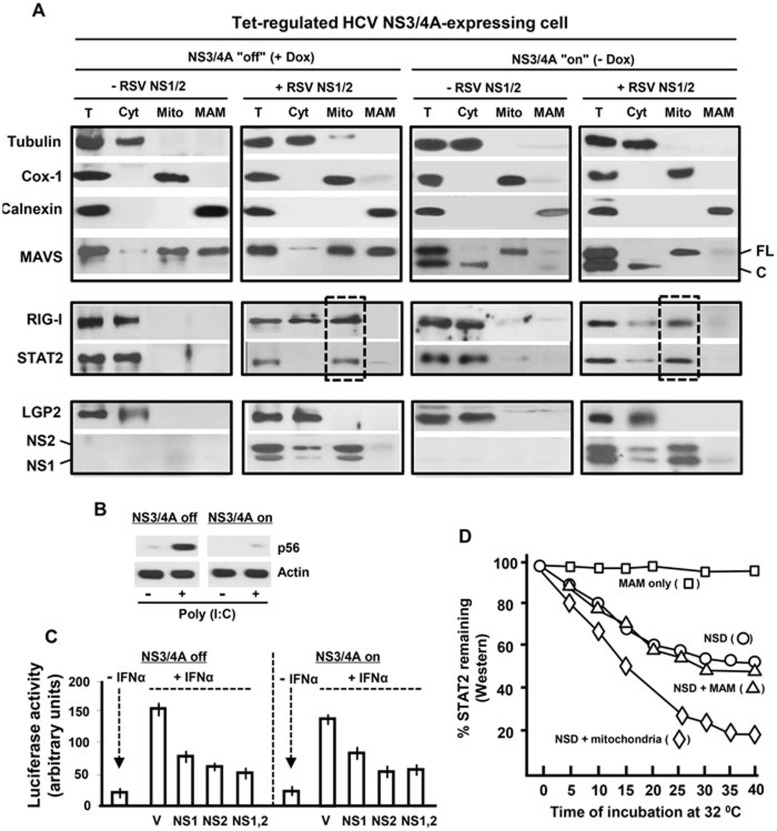
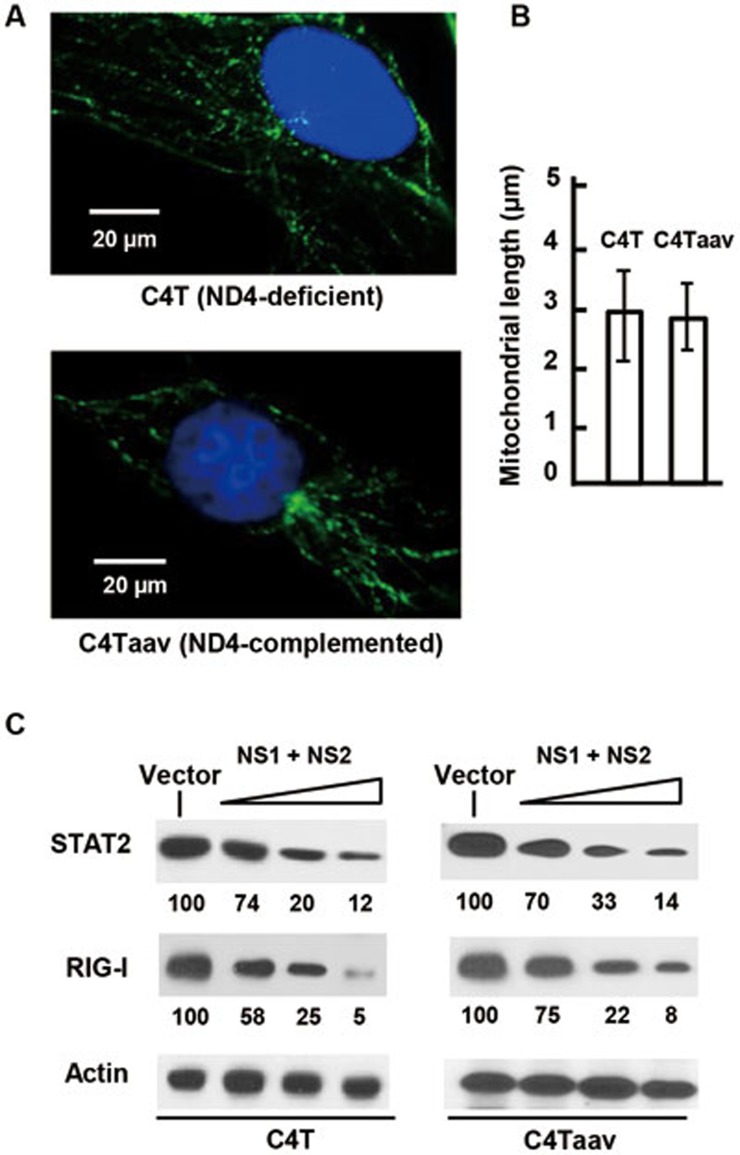
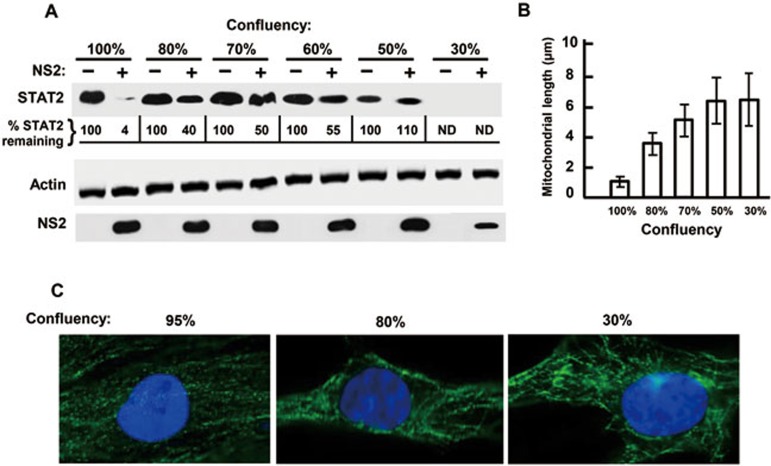
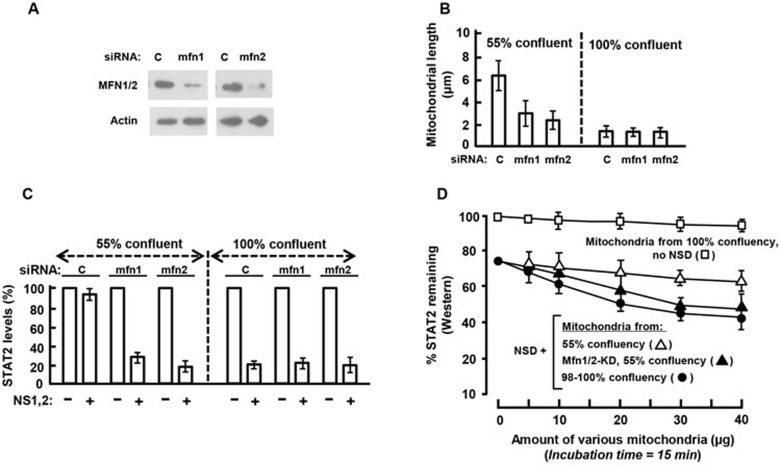
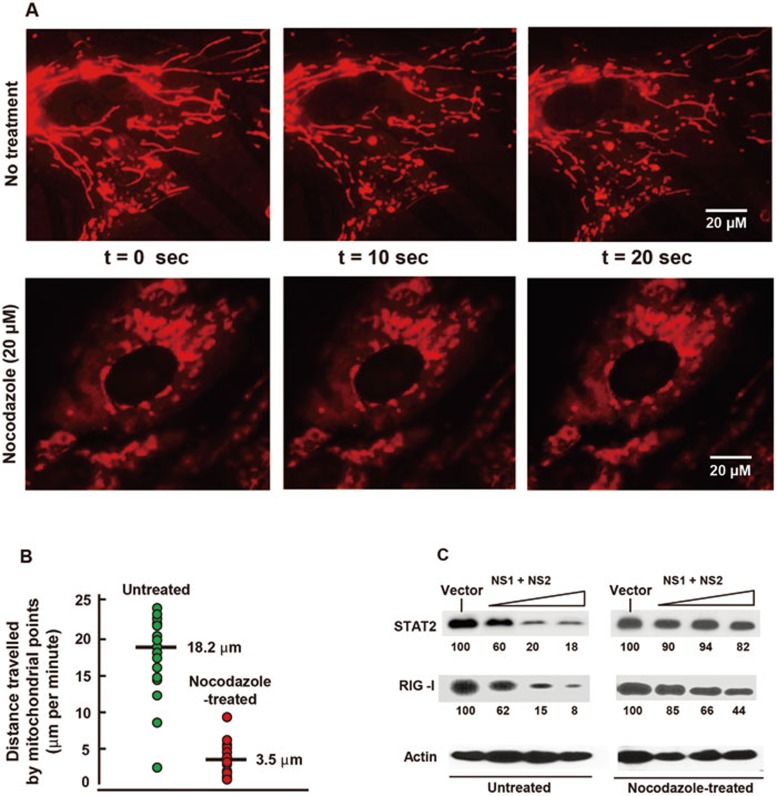
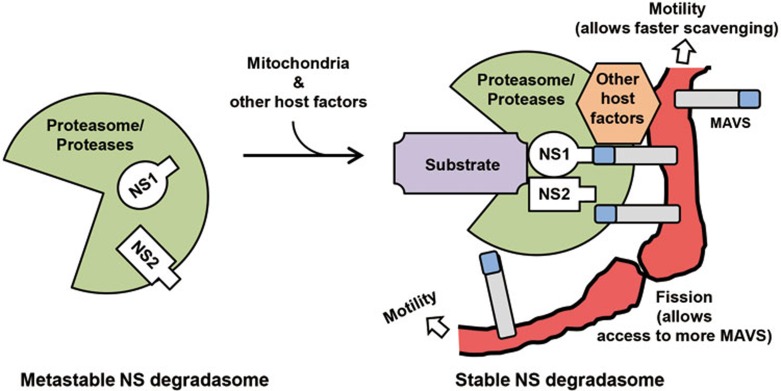
The balance between the innate immunity of the host and the ability of a pathogen to evade it strongly influences pathogenesis and virulence. The two nonstructural (NS) proteins, NS1 and NS2, of respiratory syncytial virus (RSV) are critically required for RSV virulence. Together, they strongly suppress the type I interferon (IFN)-mediated innate immunity of the host cells by degrading or inhibiting multiple cellular factors required for either IFN induction or response pathways, including RIG-I, IRF3, IRF7, TBK1 and STAT2. Here, we provide evidence for the existence of a large and heterogeneous degradative complex assembled by the NS proteins, which we named "NS-degradasome" (NSD). The NSD is roughly ∼300-750 kD in size, and its degradative activity was enhanced by the addition of purified mitochondria in vitro. Inside the cell, the majority of the NS proteins and the substrates of the NSD translocated to the mitochondria upon RSV infection. Genetic and pharmacological evidence shows that optimal suppression of innate immunity requires mitochondrial MAVS and mitochondrial motility. Together, we propose a novel paradigm in which the mitochondria, known to be important for the innate immune activation of the host, are also important for viral suppression of the innate immunity.
Figures
Similar articles
-
Respiratory syncytial virus nonstructural proteins 1 and 2: Exceptional disrupters of innate immune responses.PLoS Pathog. 2019 Oct 17;15(10):e1007984. doi: 10.1371/journal.ppat.1007984. eCollection 2019 Oct. PLoS Pathog. 2019. PMID: 31622448 Free PMC article. Review.
-
Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction.Virology. 2006 Jan 20;344(2):328-39. doi: 10.1016/j.virol.2005.09.009. Epub 2005 Oct 10. Virology. 2006. PMID: 16216295
-
Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection.PLoS One. 2012;7(2):e29386. doi: 10.1371/journal.pone.0029386. Epub 2012 Feb 27. PLoS One. 2012. PMID: 22383950 Free PMC article.
-
Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase.J Virol. 2007 Apr;81(7):3428-36. doi: 10.1128/JVI.02303-06. Epub 2007 Jan 24. J Virol. 2007. PMID: 17251292 Free PMC article.
-
Antiviral innate immunity pathways.Cell Res. 2006 Feb;16(2):141-7. doi: 10.1038/sj.cr.7310019. Cell Res. 2006. PMID: 16474426 Review.
Cited by
-
An Unexpected Encounter: Respiratory Syncytial Virus Nonstructural Protein 1 Interacts with Mediator Subunit MED25.J Virol. 2022 Oct 12;96(19):e0129722. doi: 10.1128/jvi.01297-22. Epub 2022 Sep 14. J Virol. 2022. PMID: 36102648 Free PMC article. Review.
-
Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission.Brain. 2015 Oct;138(Pt 10):2834-46. doi: 10.1093/brain/awv182. Epub 2015 Jun 29. Brain. 2015. PMID: 26122121 Free PMC article.
-
The nonstructural proteins of Pneumoviruses are remarkably distinct in substrate diversity and specificity.Virol J. 2017 Nov 6;14(1):215. doi: 10.1186/s12985-017-0881-7. Virol J. 2017. PMID: 29110727 Free PMC article.
-
Opposite Responses of Interferon and Proinflammatory Cytokines Induced by Human Metapneumovirus and Respiratory Syncytial Virus in Macrophages.Pathogens. 2025 Jul 14;14(7):694. doi: 10.3390/pathogens14070694. Pathogens. 2025. PMID: 40732740 Free PMC article.
-
Molecular Mechanisms of Innate Immune Inhibition by Non-Segmented Negative-Sense RNA Viruses.J Mol Biol. 2016 Aug 28;428(17):3467-82. doi: 10.1016/j.jmb.2016.07.017. Epub 2016 Jul 31. J Mol Biol. 2016. PMID: 27487481 Free PMC article. Review.
References
-
- Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. - PubMed
-
- Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. - PubMed
-
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF3. Cell. 2005;122:669–682. - PubMed
-
- Xu LG, Wang YY, Han KJ, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. - PubMed
-
- Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol. 2011;23:564–572. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
