

Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis
- PMID: 23881705
- PMCID: PMC4126425
- DOI: 10.1007/s11481-013-9489-x
Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis
Abstract
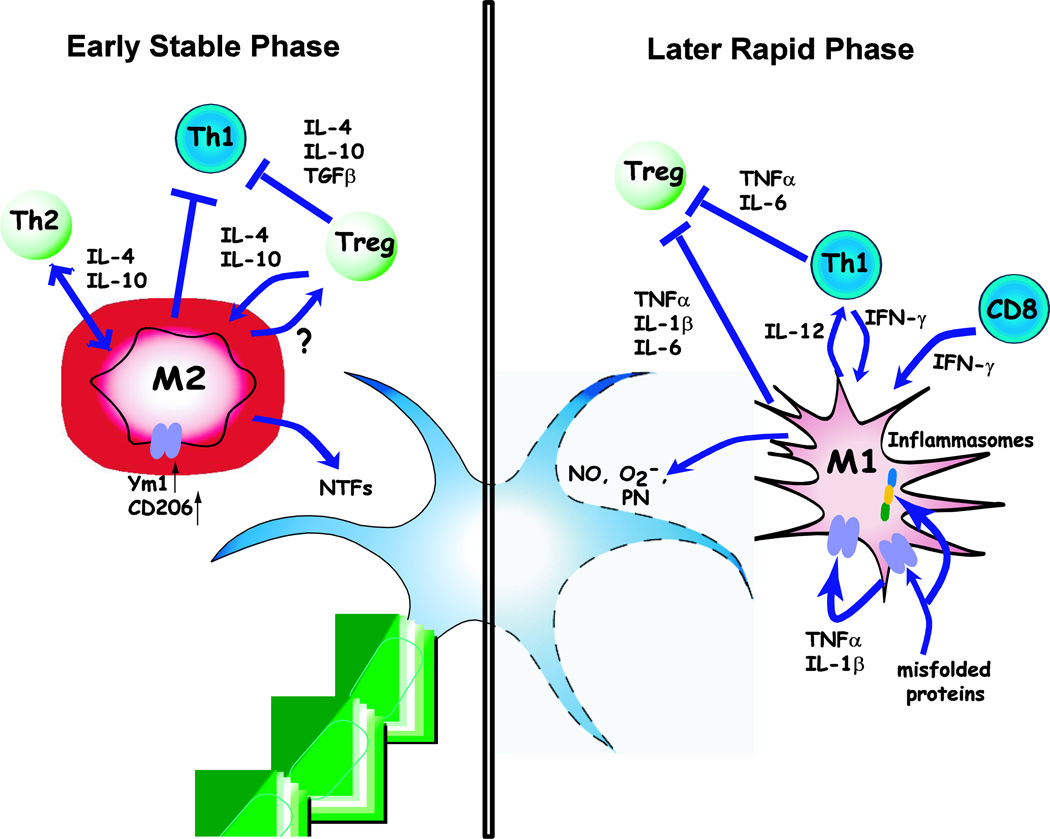
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease with selective loss of upper and lower motor neurons. At sites of motor neuron injury, neuroinflammation is a prominent pathological finding and is characterized by microglial activation, astrogliosis, and infiltration of monocytes and T-cells. Both innate and adaptive immune responses actively influence disease progression in animal models and in ALS patients, and promote neuroprotection or neurotoxicity at different stages of disease. The early immune reaction to signals from injured motor neurons is to rescue and repair damaged tissue. As disease accelerates, a shift occurs from beneficial immune responses (involving M2 microglia and regulatory T-cells) to deleterious immune responses (involving M1 microglia and Th1 cells). In this review, we underscore the importance of immune-mediated mechanisms in the pathogenesis of ALS and discuss the alterations and distinct phenotypes of immune cells at the different stages of disease. The better we understand the dynamic changes that occur within the immune system over the course of disease, the better we will be able to develop effective therapeutic regimens in ALS.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures



References
-
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
