

KIT signaling governs differential sensitivity of mature and primitive CML progenitors to tyrosine kinase inhibitors
- PMID: 23887971
- PMCID: PMC3894913
- DOI: 10.1158/0008-5472.CAN-13-1318
KIT signaling governs differential sensitivity of mature and primitive CML progenitors to tyrosine kinase inhibitors
Abstract
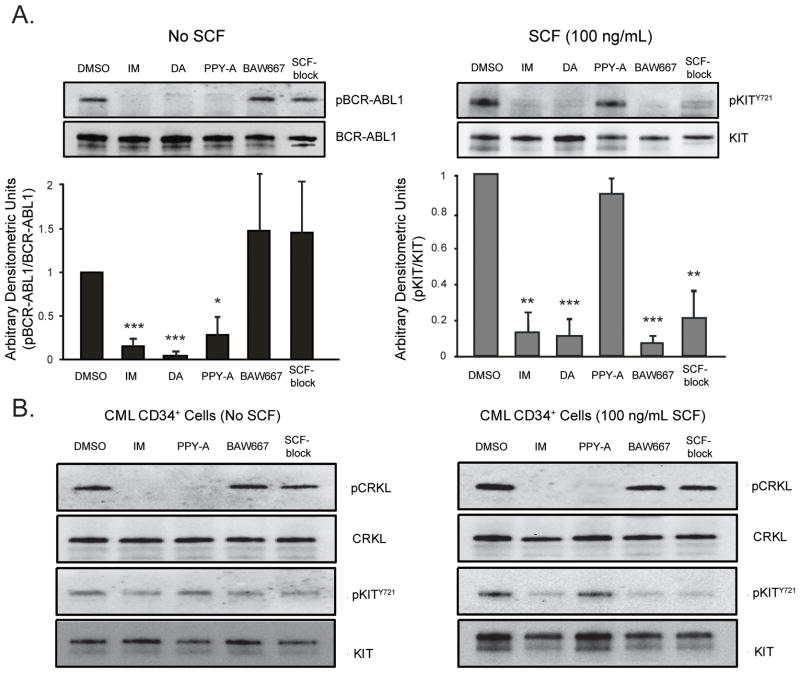
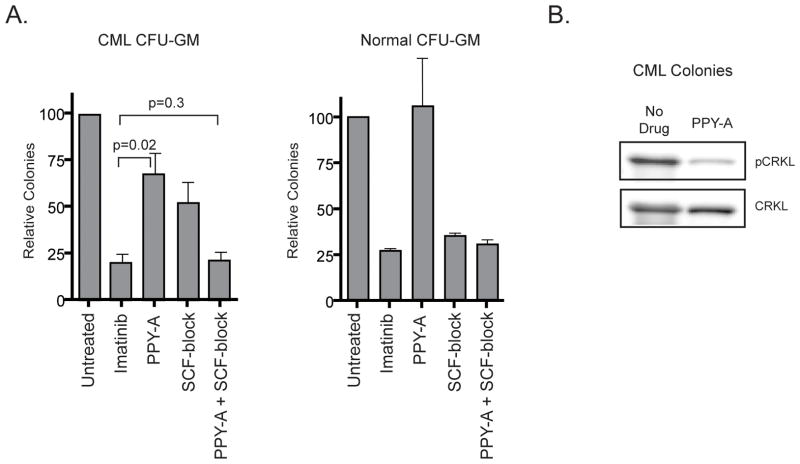
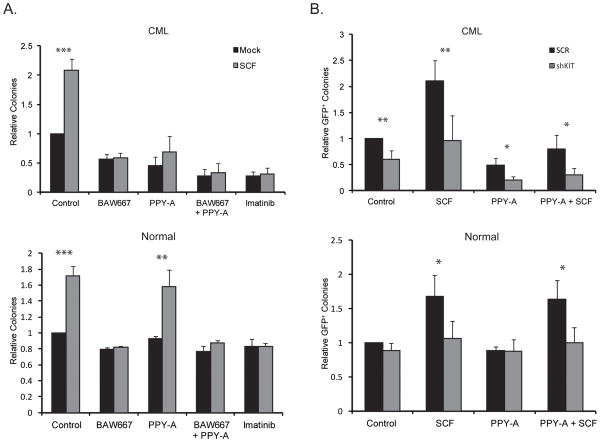
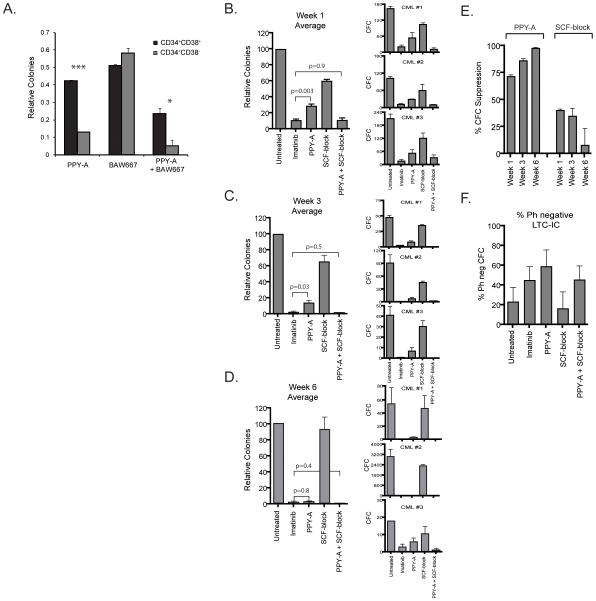
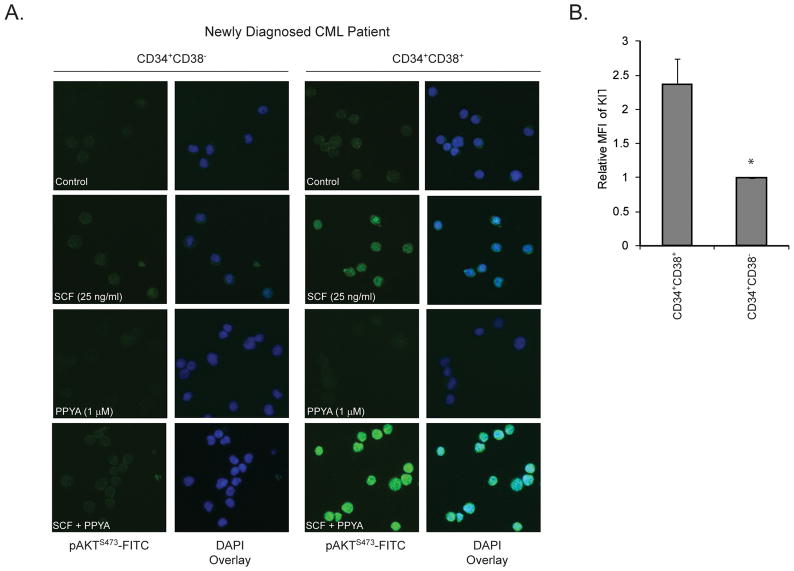
Imatinib and other BCR-ABL1 inhibitors are effective therapies for chronic myelogenous leukemia (CML), but these inhibitors target additional kinases including KIT, raising the question of whether off-target effects contribute to clinical efficacy. On the basis of its involvement in CML pathogenesis, we hypothesized that KIT may govern responses of CML cells to imatinib. To test this, we assessed the growth of primary CML progenitor cells under conditions of sole BCR-ABL1, sole KIT, and dual BCR-ABL1/KIT inhibition. Sole BCR-ABL1 inhibition suppressed mature CML progenitor cells, but these effects were largely abolished by stem cell factor (SCF) and maximal suppression required dual BCR-ABL1/KIT inhibition. In contrast, KIT inhibition did not add to the effects of BCR-ABL1 inhibition in primitive progenitors, represented by CD34(+)38(-) cells. Long-term culture-initiating cell assays on murine stroma revealed profound depletion of primitive CML cells by sole BCR-ABL1 inhibition despite the presence of SCF, suggesting that primitive CML cells are unable to use SCF as a survival factor upon BCR-ABL1 inhibition. In CD34(+)38(+) cells, SCF strongly induced pAKT(S473) in a phosphoinositide 3-kinase (PI3K)-dependent manner, which was further enhanced by inhibition of BCR-ABL1 and associated with increased colony survival. In contrast, pAKT(S473) levels remained low in CD34(+)38(-) cells cultured under the same conditions. Consistent with reduced response to SCF, KIT surface expression was significantly lower on CD34(+)38(-) compared with CD34(+)38(+) CML cells, suggesting a possible mechanism for the differential effects of SCF on mature and primitive CML progenitor cells.
©2013 AACR.
Conflict of interest statement
Disclosure of Potential Conflicts of Interest:
M.W.D. is a consultant for BMS, Novartis, ARIAD and Incyte and his laboratory receives research funding from BMS and Novartis. B.J.D. is principal investigator or co-investigator on Novartis, Bristol-Myers Squibb, and ARIAD clinical trials. His institution has contracts with these companies to pay for patient costs, nurse and data manager salaries, and institutional overhead. He does not derive salary, nor does his lab receive funds from these contracts. OHSU and B.J.D. have a financial interest in MolecularMD. OHSU has licensed technology used in some of these clinical trials to MolecularMD. This potential individual and institutional conflict of interest has been reviewed and managed by OHSU. P.W.M. is an employee of Novartis Pharma, Basel, Switzerland. J.E.C. receives research funding from ARIAD, Novartis, BMS, Pfizer, Chemgenex, Deciphera, and Incyte.
Figures
References
-
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17. - PubMed
-
- Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–53. - PubMed
-
- Dewar AL, Cambareri AC, Zannettino AC, Miller BL, Doherty KV, Hughes TP, et al. Macrophage colony-stimulating factor receptor c-fms is a novel target of imatinib. Blood. 2005;105:3127–32. - PubMed
-
- Manley PW, Stiefl N, Cowan-Jacob SW, Kaufman S, Mestan J, Wartmann M, et al. Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg Med Chem. 2010;18:6977–86. - PubMed
-
- Al-Ali HK, Heinrich MC, Lange T, Krahl R, Mueller M, Muller C, et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patients with secondary resistance to imatinib. Hematol J. 2004;5:55–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
