

Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms
- PMID: 23897027
- PMCID: PMC4045499
- DOI: 10.1007/s00401-013-1115-8
Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms
Abstract
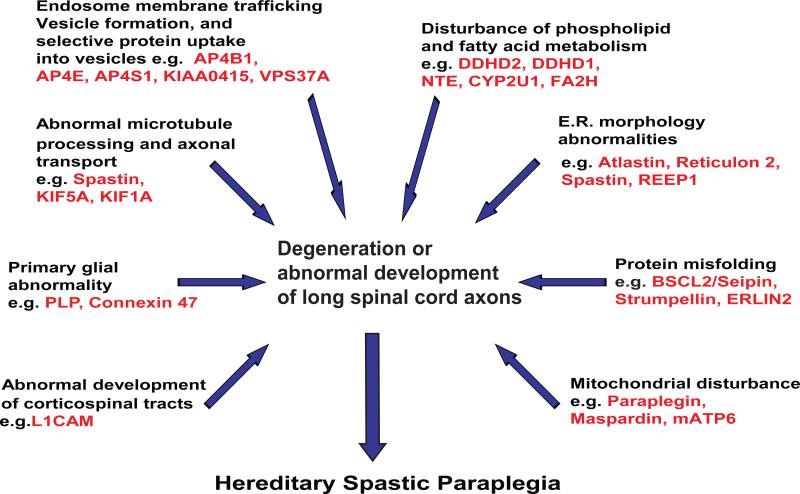
Hereditary spastic paraplegia (HSP) is a syndrome designation describing inherited disorders in which lower extremity weakness and spasticity are the predominant symptoms. There are more than 50 genetic types of HSP. HSP affects individuals of diverse ethnic groups with prevalence estimates ranging from 1.2 to 9.6 per 100,000. Symptoms may begin at any age. Gait impairment that begins after childhood usually worsens very slowly over many years. Gait impairment that begins in infancy and early childhood may not worsen significantly. Postmortem studies consistently identify degeneration of corticospinal tract axons (maximal in the thoracic spinal cord) and degeneration of fasciculus gracilis fibers (maximal in the cervico-medullary region). HSP syndromes thus appear to involve motor-sensory axon degeneration affecting predominantly (but not exclusively) the distal ends of long central nervous system (CNS) axons. In general, proteins encoded by HSP genes have diverse functions including (1) axon transport (e.g. SPG30/KIF1A, SPG10/KIF5A and possibly SPG4/Spastin); (2) endoplasmic reticulum morphology (e.g. SPG3A/Atlastin, SPG4/Spastin, SPG12/reticulon 2, and SPG31/REEP1, all of which interact); (3) mitochondrial function (e.g. SPG13/chaperonin 60/heat-shock protein 60, SPG7/paraplegin; and mitochondrial ATP6); (4) myelin formation (e.g. SPG2/Proteolipid protein and SPG42/Connexin 47); (5) protein folding and ER-stress response (SPG6/NIPA1, SPG8/K1AA0196 (Strumpellin), SGP17/BSCL2 (Seipin), "mutilating sensory neuropathy with spastic paraplegia" owing to CcT5 mutation and presumably SPG18/ERLIN2); (6) corticospinal tract and other neurodevelopment (e.g. SPG1/L1 cell adhesion molecule and SPG22/thyroid transporter MCT8); (7) fatty acid and phospholipid metabolism (e.g. SPG28/DDHD1, SPG35/FA2H, SPG39/NTE, SPG54/DDHD2, and SPG56/CYP2U1); and (8) endosome membrane trafficking and vesicle formation (e.g. SPG47/AP4B1, SPG48/KIAA0415, SPG50/AP4M1, SPG51/AP4E, SPG52/AP4S1, and VSPG53/VPS37A). The availability of animal models (including bovine, murine, zebrafish, Drosophila, and C. elegans) for many types of HSP permits exploration of disease mechanisms and potential treatments. This review highlights emerging concepts of this large group of clinically similar disorders.
Figures
References
-
- Abou-Donia MB. Organophosphorus ester-induced delayed neurotoxicity. Ann Rev Pharmacol Toxicol. 1981;21:511–548. - PubMed
-
- Ahmed FE, Qureshi IM, Wooldridge MAW, Pejaver RK. Hereditary spastic paraplegia and Evans's syndrome. Acta Paediat. 1996;85:879–881. - PubMed
-
- Al-Saif A, Bohlega S, Al-Mohanna F. Loss of ERLIN2 function leads to juvenile primary lateral sclerosis. Ann Neurol. 2012;72:510–516. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
