

Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome
- PMID: 23897642
- PMCID: PMC3829633
- DOI: 10.1002/ajmg.a.36044
Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome
Abstract
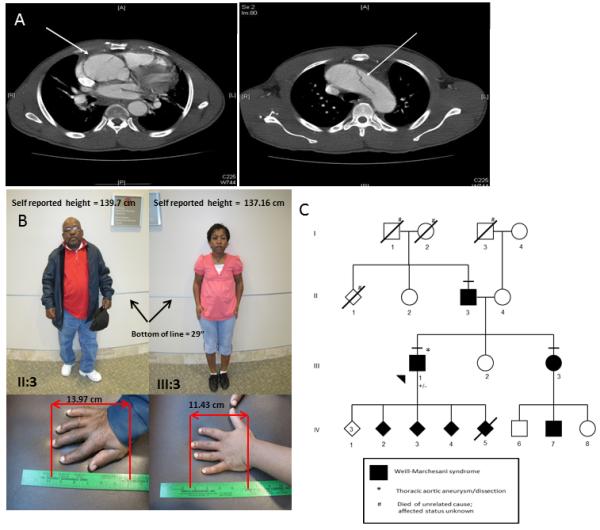
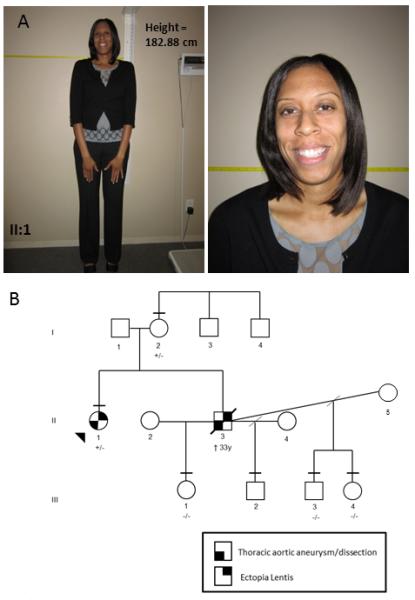
Mutations in FBN1 cause a range of overlapping but distinct conditions including Marfan syndrome (MFS), Weill-Marchesani syndrome (WMS), familial thoracic aortic aneurysms/dissections (FTAAD), acromicric dysplasia (AD), and geleophysic dysplasia (GD). Two forms of acromelic dysplasia, AD and GD, characterized by short stature, brachydactyly, reduced joint mobility, and characteristic facies, result from heterozygous missense mutations occurring in exons 41 and 42 of FBN1; missense mutations in these exons have not been reported to cause MFS or other syndromes. Here we report on probands with MFS and WMS who have heterozygous FBN1 missense mutations in exons 41 and 42, respectively. The proband with WMS has ectopia lentis, short stature, thickened pinnae, tight skin, striae atrophicae, reduced extension of the elbows, contractures of the fingers and toes, and brachydactyly and has a missense mutation in exon 42 of FBN1 (c.5242T>C; p.C1748R). He also experienced a previously unreported complication of WMS, an acute thoracic aortic dissection. The second proband displays classic characteristics of MFS, including ectopia lentis, skeletal features, and aortic root dilatation, and has a missense mutation in exon 41 of FBN1 (c.5084G>A; p.C1695Y). These phenotypes provide evidence that missense mutations in exons 41 and 42 of FBN1 lead to MFS and WMS in addition to AD and GD and also suggest that all individuals with pathogenic FBN1 mutations in these exons should be assessed for thoracic aortic disease and ectopia lentis. Further studies are necessary to elucidate the factors responsible for the different phenotypes associated with missense mutations in these exons of FBN1.
Keywords: acromicric dysplasia; aortic aneurysm; aortic dissection; ectopia lentis; geleophysic dysplasia.
Copyright © 2013 Wiley Periodicals, Inc.
Figures
References
-
- Ades LC, Holman KJ, Brett MS, Edwards MJ, Bennetts B. Ectopia lentis phenotypes and the FBN1 gene. Am J Med Genet A. 2004;126:284–289. - PubMed
-
- Bowers D. Marfan’s syndrome and the Weill-Marchesani syndrome in the S. family. Ann Intern Med. 1959;51:1049–1070. - PubMed
-
- Evereklioglu C, Hepsen IF, Hamdi ER. Weill-Marchesani syndrome in three generations. Eye (Lond) 1999;13(Pt 6):773–777. - PubMed
-
- Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, rslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Ades LC, Biggin A, Benetts B, Brett M, Holman KJ, De BJ, Coucke P, Francke U, De PA, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
