

Reactive oxygen species in pulmonary vascular remodeling
- PMID: 23897679
- PMCID: PMC3907081
- DOI: 10.1002/cphy.c120024
Reactive oxygen species in pulmonary vascular remodeling
Abstract
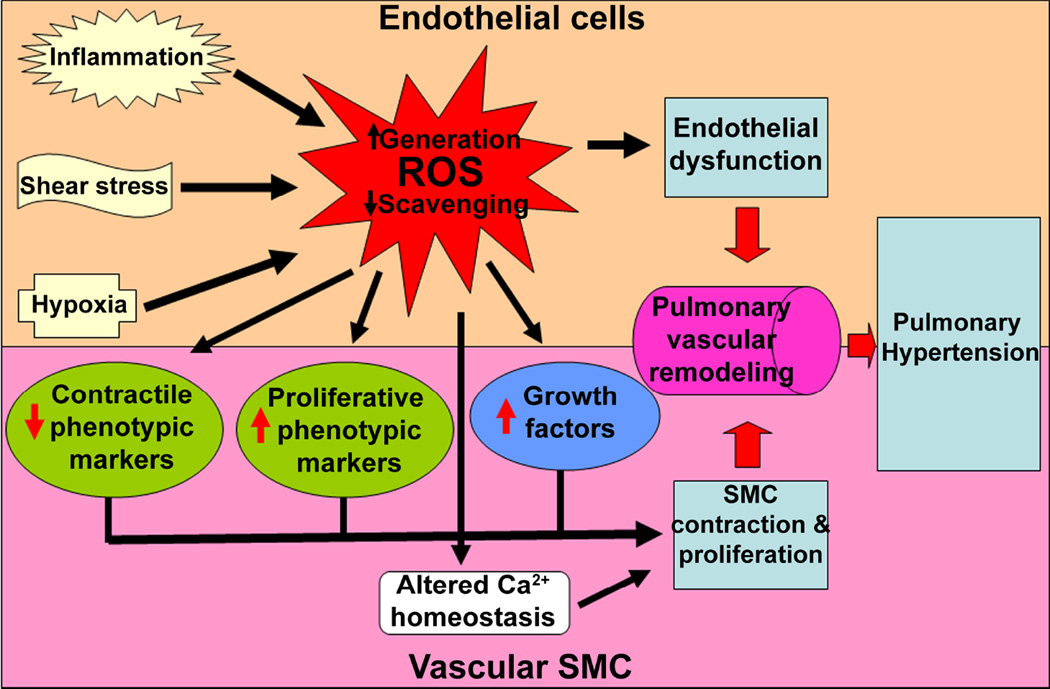
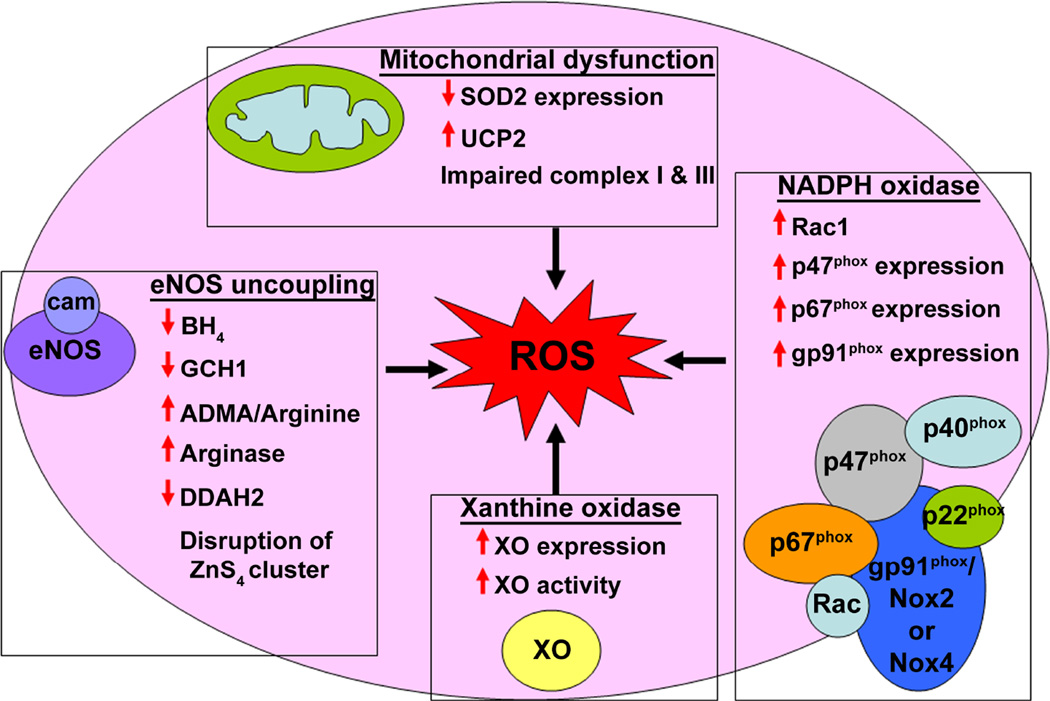
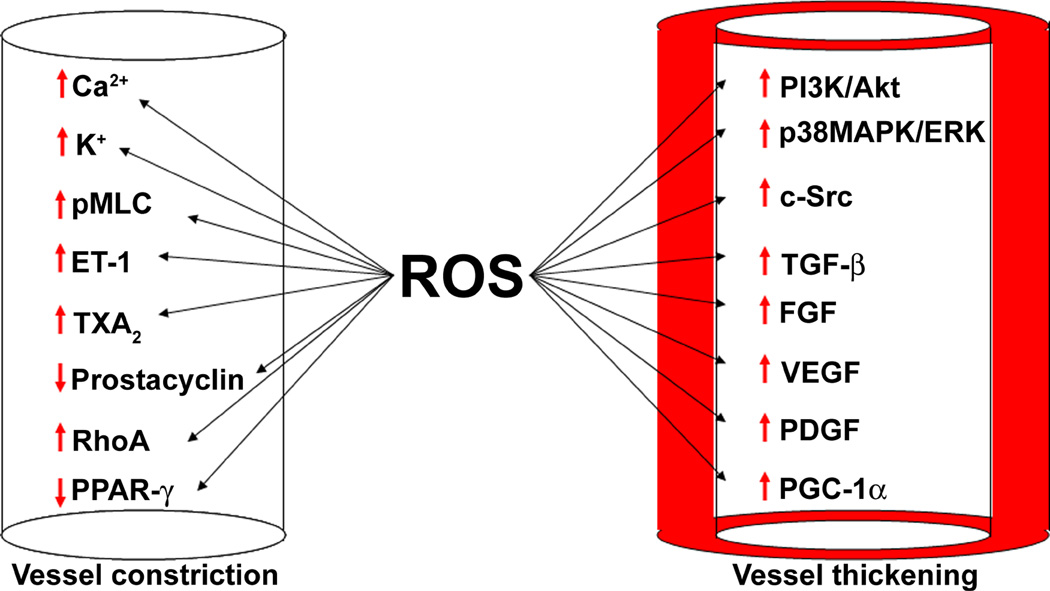
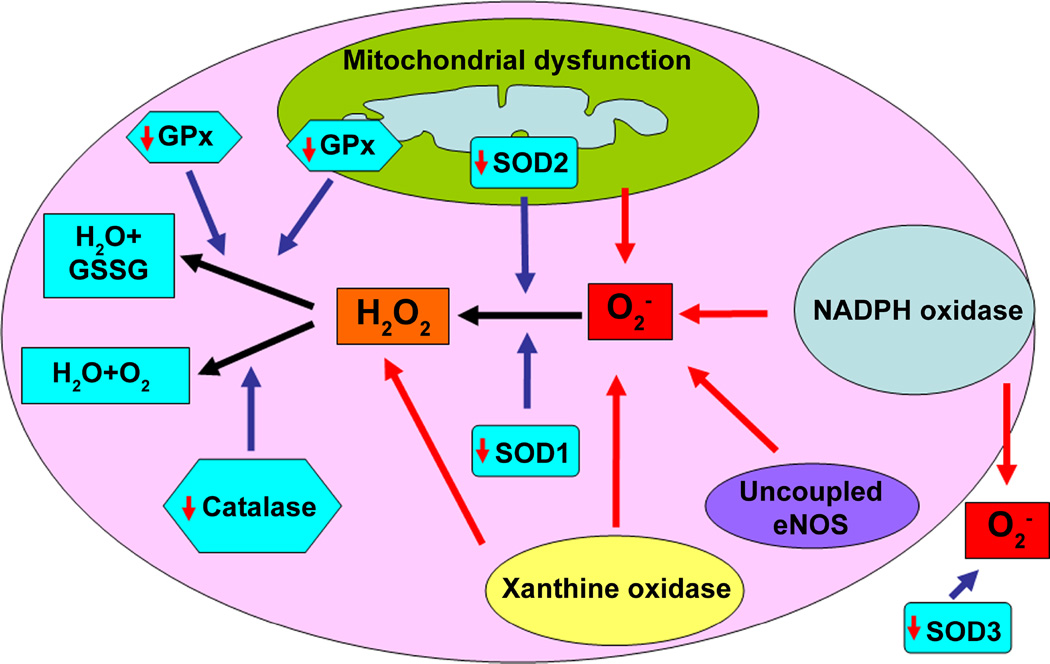
The pathogenesis of pulmonary hypertension is a complex multifactorial process that involves the remodeling of pulmonary arteries. This remodeling process encompasses concentric medial thickening of small arterioles, neomuscularization of previously nonmuscular capillary-like vessels, and structural wall changes in larger pulmonary arteries. The pulmonary arterial muscularization is characterized by vascular smooth muscle cell hyperplasia and hypertrophy. In addition, in uncontrolled pulmonary hypertension, the clonal expansion of apoptosis-resistant endothelial cells leads to the formation of plexiform lesions. Based upon a large number of studies in animal models, the three major stimuli that drive the vascular remodeling process are inflammation, shear stress, and hypoxia. Although, the precise mechanisms by which these stimuli impair pulmonary vascular function and structure are unknown, reactive oxygen species (ROS)-mediated oxidative damage appears to play an important role. ROS are highly reactive due to their unpaired valence shell electron. Oxidative damage occurs when the production of ROS exceeds the quenching capacity of the antioxidant mechanisms of the cell. ROS can be produced from complexes in the cell membrane (nicotinamide adenine dinucleotide phosphate-oxidase), cellular organelles (peroxisomes and mitochondria), and in the cytoplasm (xanthine oxidase). Furthermore, low levels of tetrahydrobiopterin (BH4) and L-arginine the rate limiting cofactor and substrate for endothelial nitric oxide synthase (eNOS), can cause the uncoupling of eNOS, resulting in decreased NO production and increased ROS production. This review will focus on the ROS generation systems, scavenger antioxidants, and oxidative stress associated alterations in vascular remodeling in pulmonary hypertension.
© 2013 American Physiological Society.
Figures
References
-
- Gaine SP, Rubin LJ. Lancet. 1998;352(9129):719–725. - PubMed
-
- Cool CD, Groshong SD, Oakey J, Voelkel NF. Chest. 2005;128(6 Suppl):565S–571S. - PubMed
-
- Meyrick B, Reid L. Laboratory investigation; a journal of technical methods and pathology. 1980;42(6):603–615. - PubMed
-
- Meyrick B, Reid L. Laboratory investigation; a journal of technical methods and pathology. 1978;38(2):188–200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
