

Bile acid metabolism and signaling
- PMID: 23897684
- PMCID: PMC4422175
- DOI: 10.1002/cphy.c120023
Bile acid metabolism and signaling
Abstract
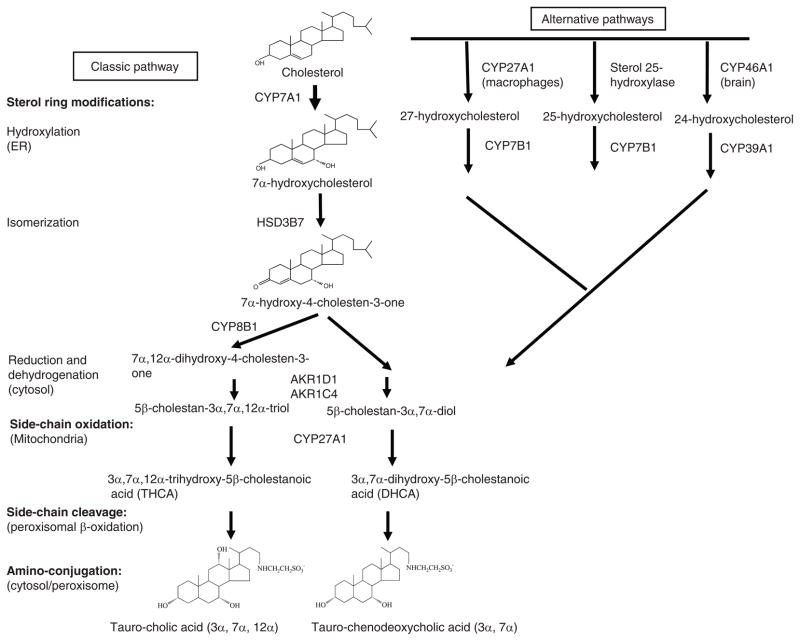
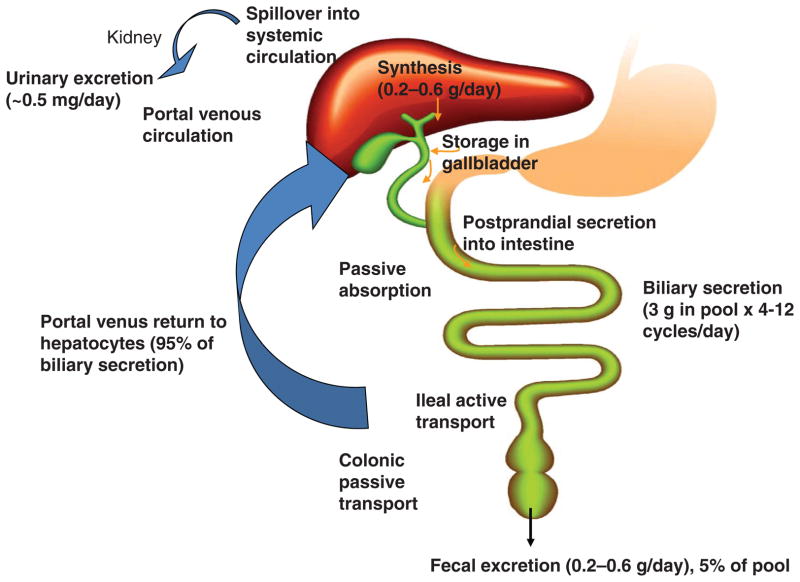
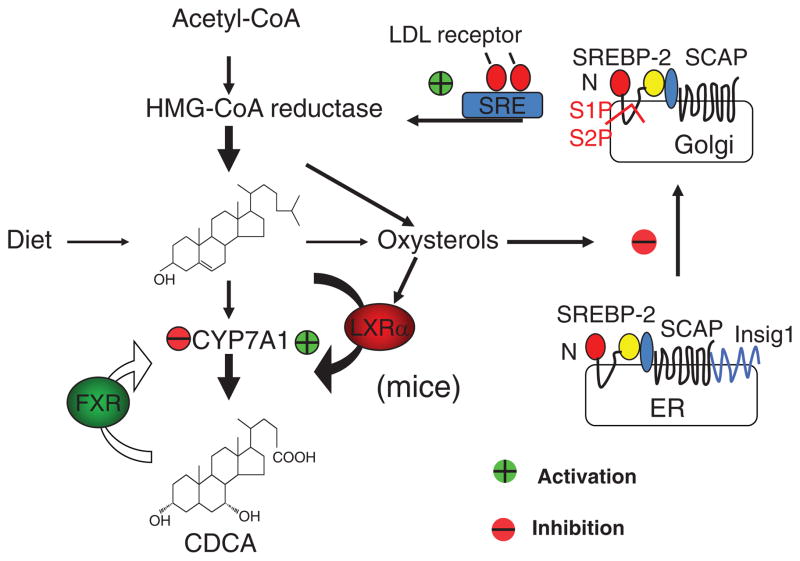
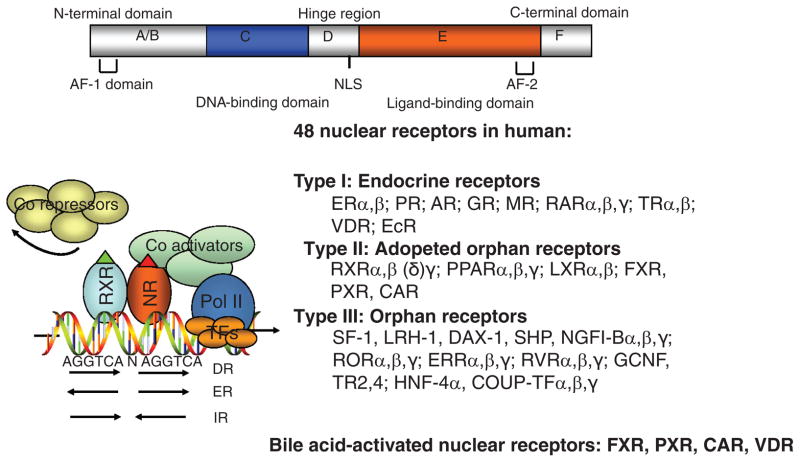
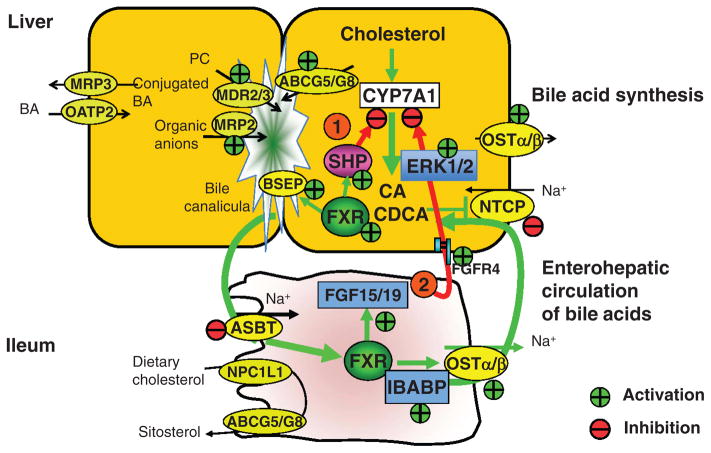
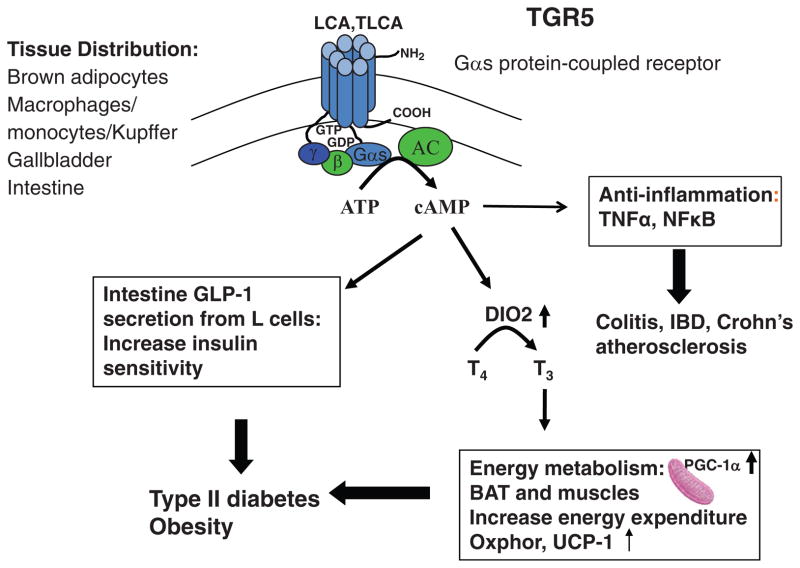
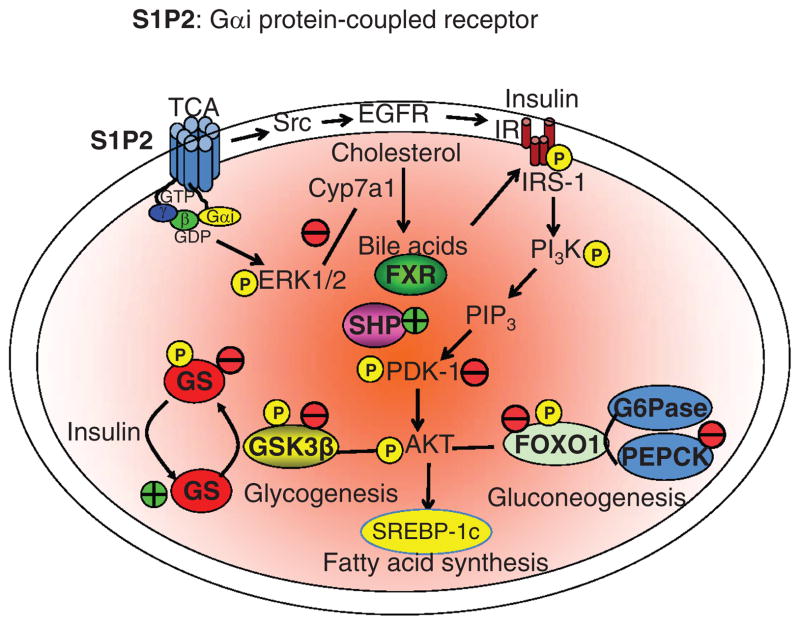
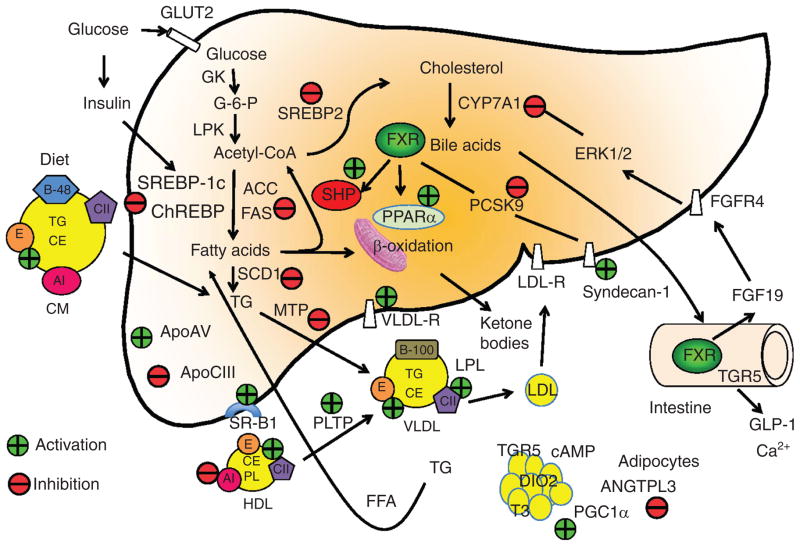
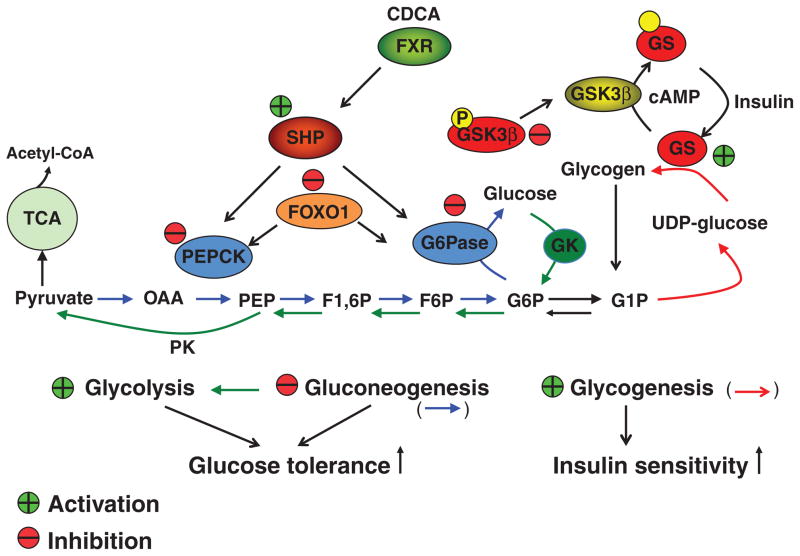
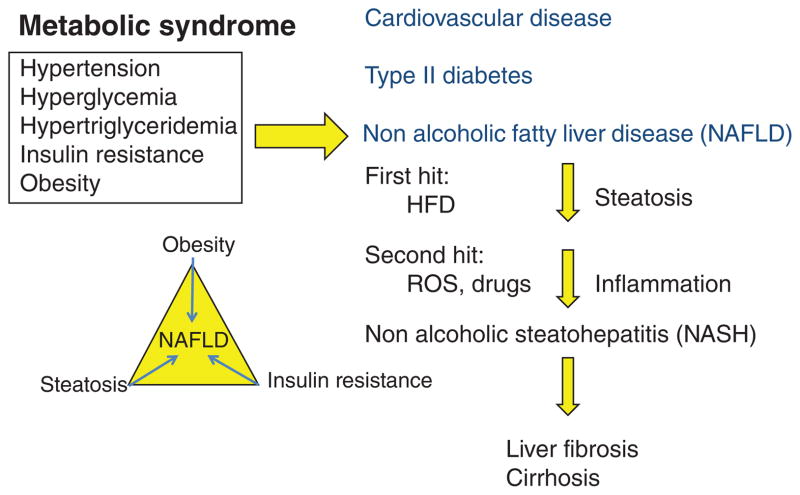
Bile acids are important physiological agents for intestinal nutrient absorption and biliary secretion of lipids, toxic metabolites, and xenobiotics. Bile acids also are signaling molecules and metabolic regulators that activate nuclear receptors and G protein-coupled receptor (GPCR) signaling to regulate hepatic lipid, glucose, and energy homeostasis and maintain metabolic homeostasis. Conversion of cholesterol to bile acids is critical for maintaining cholesterol homeostasis and preventing accumulation of cholesterol, triglycerides, and toxic metabolites, and injury in the liver and other organs. Enterohepatic circulation of bile acids from the liver to intestine and back to the liver plays a central role in nutrient absorption and distribution, and metabolic regulation and homeostasis. This physiological process is regulated by a complex membrane transport system in the liver and intestine regulated by nuclear receptors. Toxic bile acids may cause inflammation, apoptosis, and cell death. On the other hand, bile acid-activated nuclear and GPCR signaling protects against inflammation in liver, intestine, and macrophages. Disorders in bile acid metabolism cause cholestatic liver diseases, dyslipidemia, fatty liver diseases, cardiovascular diseases, and diabetes. Bile acids, bile acid derivatives, and bile acid sequestrants are therapeutic agents for treating chronic liver diseases, obesity, and diabetes in humans.
© 2013 American Physiological Society.
Figures
References
-
- Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today. 2012;17:988–997. - PubMed
-
- Aldridge MA, Ito MK. Colesevelam hydrochloride: A novel bile acid-binding resin. Ann Pharmacother. 2001;35:898–907. - PubMed
-
- Alvarez L, Jara P, Sanchez-Sabate E, Hierro L, Larrauri J, Diaz MC, Camarena C, De La Vega A, Frauca E, Lopez-Collazo E, Lapunzina P. Reduced hepatic expression of Farnesoid X Receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13:2451–2460. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
