

Release of targeted p53 from the mitochondrion as an early signal during mitochondrial dysfunction
- PMID: 23899557
- PMCID: PMC3826263
- DOI: 10.1016/j.cellsig.2013.07.019
Release of targeted p53 from the mitochondrion as an early signal during mitochondrial dysfunction
Abstract
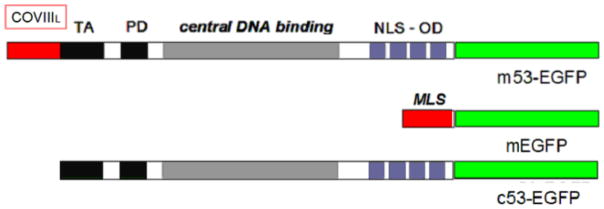
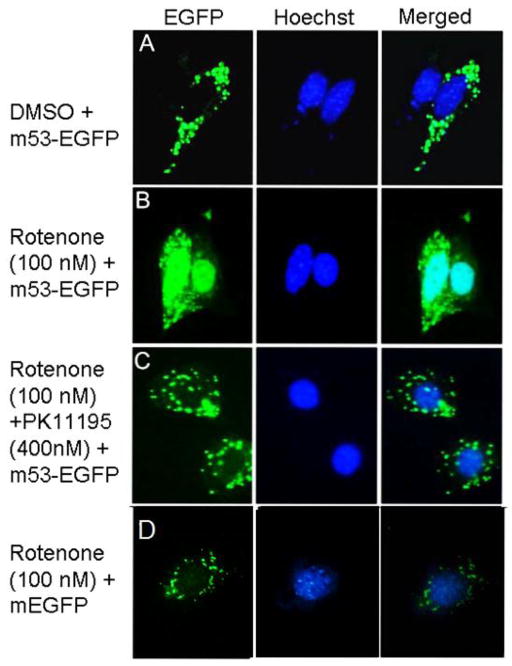
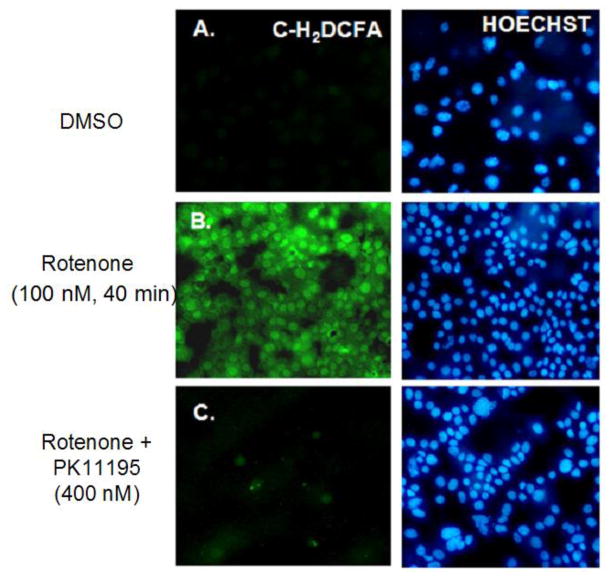
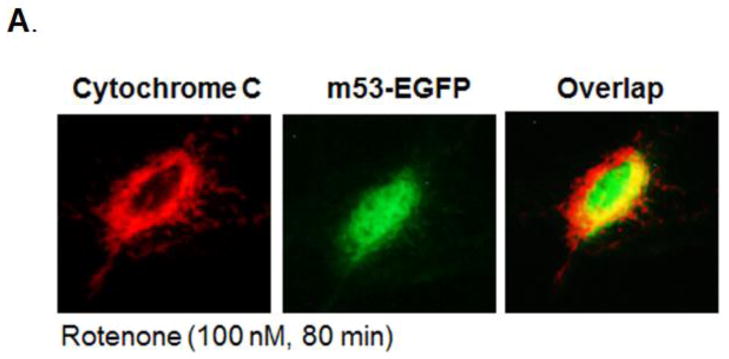
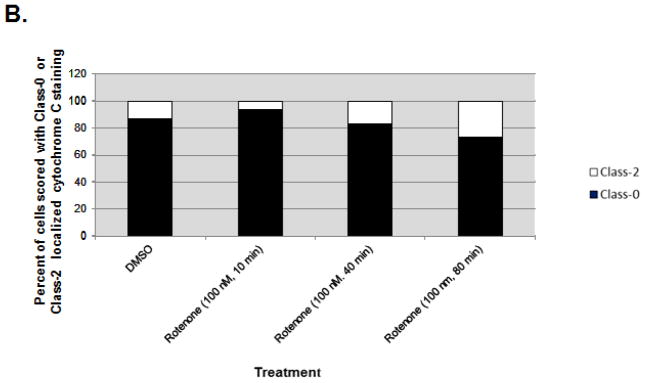
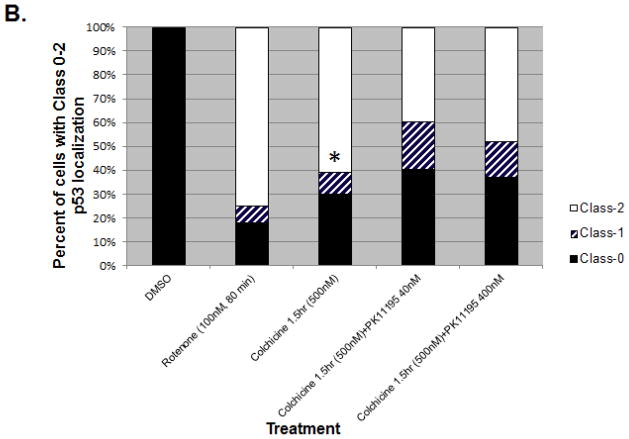
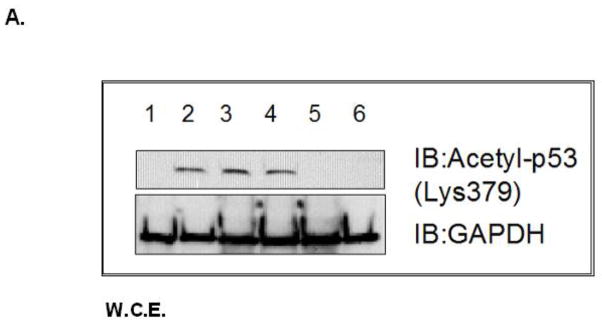
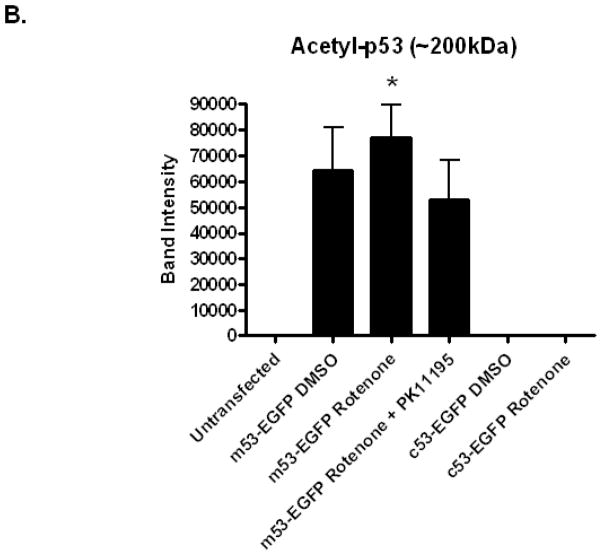
Increased accumulation of p53 tumor suppressor protein is an early response to low-level stressors. To investigate the fate of mitochondrial-sequestered p53, mouse embryonic fibroblast cells (MEFs) on a p53-deficient genetic background were transfected with p53-EGFP fusion protein led by a sense (m53-EGFP) or antisense (c53-EGFP) mitochondrial import signal. Rotenone exposure (100nM, 1h) triggered the translocation of m53-EGFP from the mitochondrion to the nucleus, thus shifting the transfected cells from a mitochondrial p53 to a nuclear p53 state. Antibodies for p53 serine phosphorylation or lysine acetylation indicated a different post-translational status of recombinant p53 in the nucleus and mitochondrion, respectively. These data suggest that cycling of p53 through the mitochondria may establish a direct pathway for p53 signaling from the mitochondria to the nucleus during mitochondrial dysfunction. PK11195, a pharmacological ligand of mitochondrial TSPO (formerly known as the peripheral-type benzodiazepine receptor), partially suppressed the release of mitochondria-sequestered p53. These findings support the notion that p53 function mediates a direct signaling pathway from the mitochondria to nucleus during mitochondrial dysfunction.
Keywords: Embryo; Mitochondria; PK11195; Rotenone; TspO; p53.
© 2013. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures




Similar articles
-
Direct influence of the p53 tumor suppressor on mitochondrial biogenesis and function.FASEB J. 2001 Mar;15(3):635-44. doi: 10.1096/fj.00-0262com. FASEB J. 2001. PMID: 11259382
-
GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria.Cell Death Differ. 2011 May;18(5):874-86. doi: 10.1038/cdd.2010.153. Epub 2010 Dec 3. Cell Death Differ. 2011. PMID: 21127500 Free PMC article.
-
Pharmacological inhibition of GSK3 attenuates DNA damage-induced apoptosis via reduction of p53 mitochondrial translocation and Bax oligomerization in neuroblastoma SH-SY5Y cells.Cell Mol Biol Lett. 2013 Mar;18(1):58-74. doi: 10.2478/s11658-012-0039-y. Epub 2012 Nov 16. Cell Mol Biol Lett. 2013. PMID: 23161404 Free PMC article.
-
p53 regulates mitochondrial dynamics by inhibiting Drp1 translocation into mitochondria during cellular senescence.FASEB J. 2020 Feb;34(2):2451-2464. doi: 10.1096/fj.201901747RR. Epub 2019 Dec 13. FASEB J. 2020. PMID: 31908078
-
Discovery of Mieap-regulated mitochondrial quality control as a new function of tumor suppressor p53.Cancer Sci. 2017 May;108(5):809-817. doi: 10.1111/cas.13208. Epub 2017 May 5. Cancer Sci. 2017. PMID: 28222492 Free PMC article. Review.
Cited by
-
Analysis of Proapoptotic Protein Trafficking to and from Mitochondria.Methods Mol Biol. 2021;2310:161-178. doi: 10.1007/978-1-0716-1433-4_10. Methods Mol Biol. 2021. PMID: 34096003
-
Regulation of H9C2 cell hypertrophy by 14-3-3η via inhibiting glycolysis.PLoS One. 2024 Jul 22;19(7):e0307696. doi: 10.1371/journal.pone.0307696. eCollection 2024. PLoS One. 2024. PMID: 39038022 Free PMC article.
-
Protein phosphatase 2A-B55β mediated mitochondrial p-GPX4 dephosphorylation promoted sorafenib-induced ferroptosis in hepatocellular carcinoma via regulating p53 retrograde signaling.Theranostics. 2023 Jul 31;13(12):4288-4302. doi: 10.7150/thno.82132. eCollection 2023. Theranostics. 2023. PMID: 37554285 Free PMC article.
References
-
- Gasparre G, Kurelac I, Capristo M, Iommarini L, Ghelli A, Ceccarelli C, Nicoletti G, Nanni P, De Giovanni C, Scotlandi K, Betts C, Carelli V, Lollini P, Romeo G, Rugolo M, Porcelli A. Cancer Res. 2011 Oct 1;71(19):6220–6229. - PubMed
-
- Modica-Napolitano J, Kulawiec M, Singh K. Curr Mol Med. 2007 Feb;(1):121–31. Review. - PubMed
-
- Larosche I, Lettéron P, Fromenty B, Vadrot N, Abbey-Toby A, Feldmann G, Pessavre D, Mansouri A. J Pharm and Exp Thera. 2007;321(2):526–535. - PubMed
-
- Desouki M, Kulawiec M, Bansal S, Das G, Singh K. Cancer Biol Ther. 2005 Dec;4(12):1367–73. - PubMed
-
- Zhou S, Kachhap S, Singh K. Mutagenesis. 2003;18:287–292. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
