

One contact for every twelve residues allows robust and accurate topology-level protein structure modeling
- PMID: 23900763
- PMCID: PMC4128384
- DOI: 10.1002/prot.24374
One contact for every twelve residues allows robust and accurate topology-level protein structure modeling
Abstract
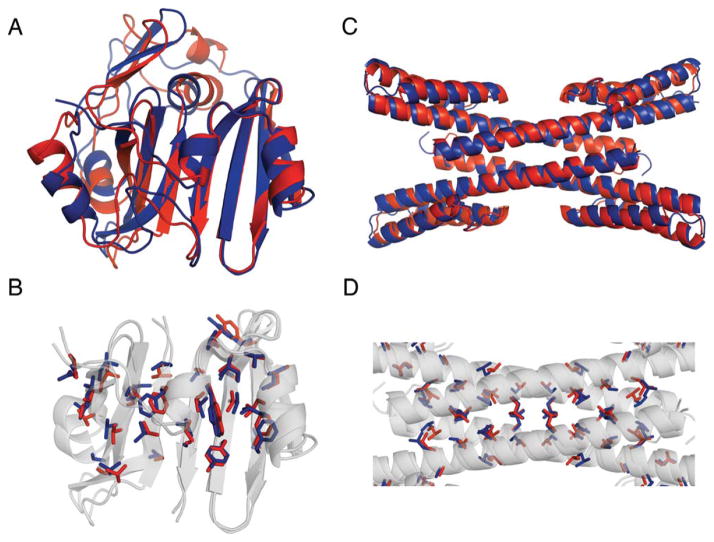
A number of methods have been described for identifying pairs of contacting residues in protein three-dimensional structures, but it is unclear how many contacts are required for accurate structure modeling. The CASP10 assisted contact experiment provided a blind test of contact guided protein structure modeling. We describe the models generated for these contact guided prediction challenges using the Rosetta structure modeling methodology. For nearly all cases, the submitted models had the correct overall topology, and in some cases, they had near atomic-level accuracy; for example the model of the 384 residue homo-oligomeric tetramer (Tc680o) had only 2.9 Å root-mean-square deviation (RMSD) from the crystal structure. Our results suggest that experimental and bioinformatic methods for obtaining contact information may need to generate only one correct contact for every 12 residues in the protein to allow accurate topology level modeling.
Keywords: ab initio prediction; comparative modeling; contact prediction; homology modeling; protein structure prediction; rosetta.
Copyright © 2013 Wiley Periodicals, Inc.
Figures
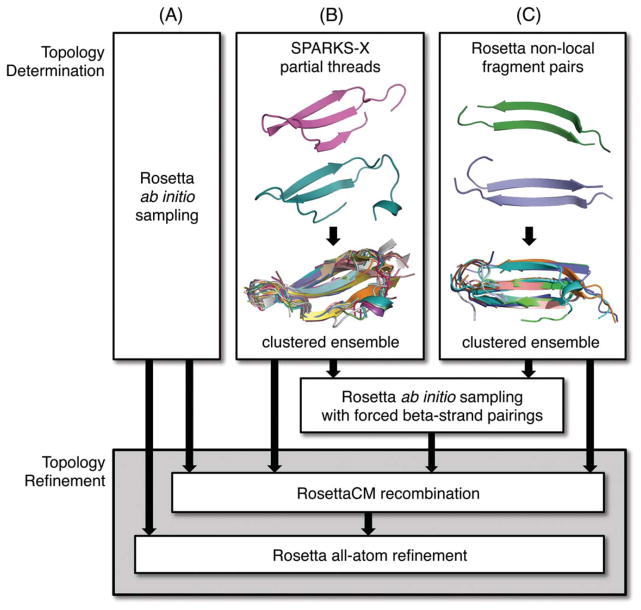
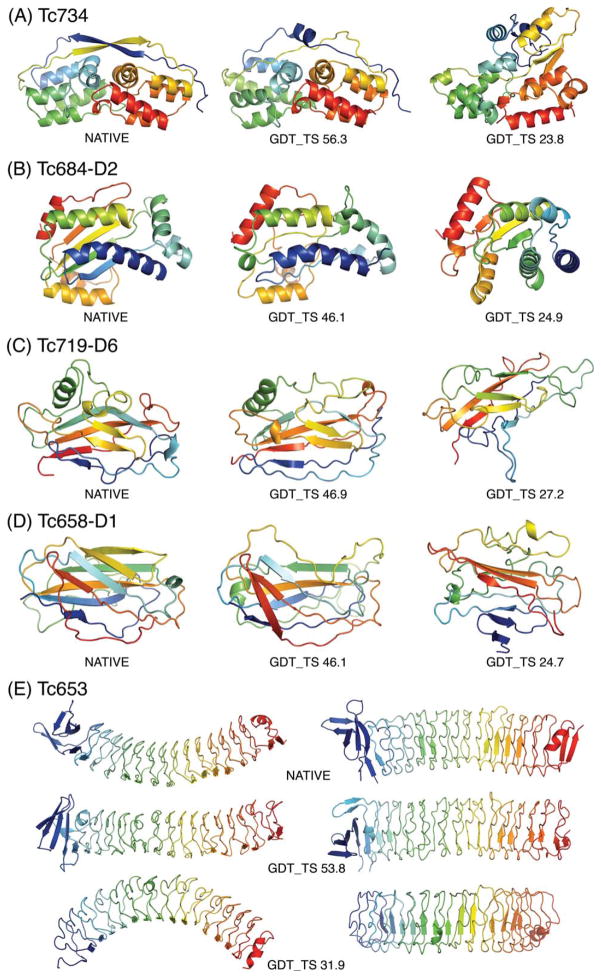
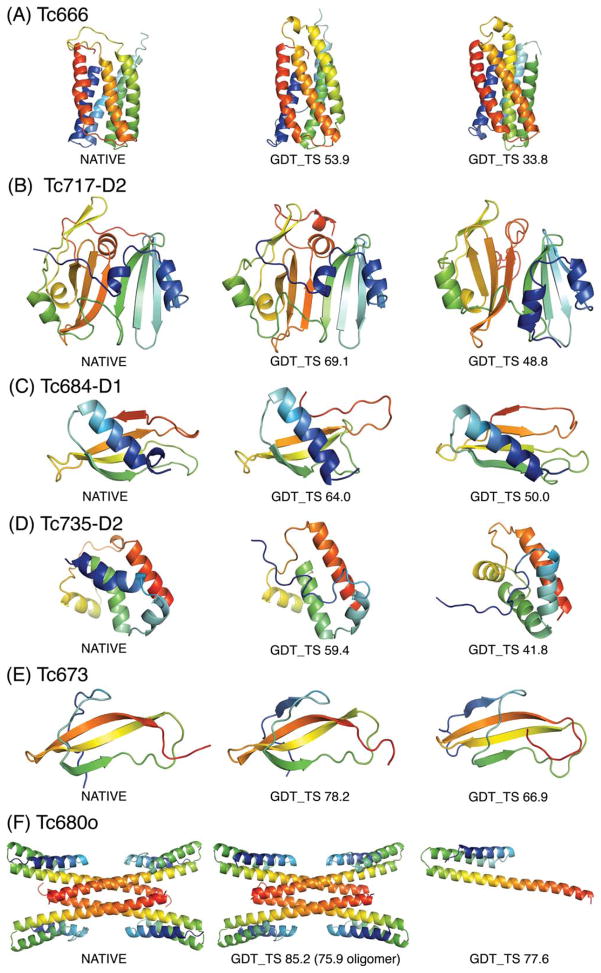
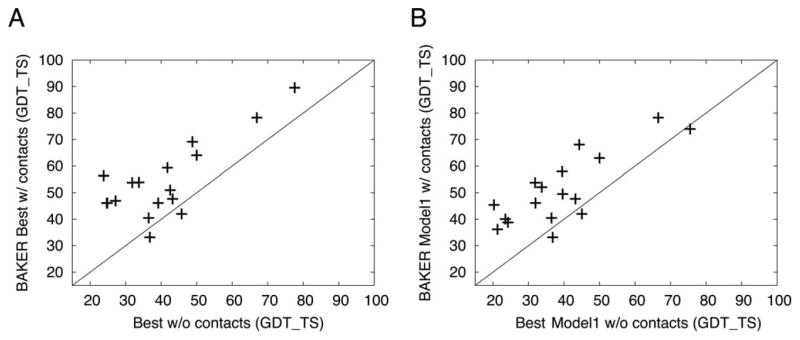
References
-
- Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. - PubMed
-
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci U S A. 2008;105:4685–4690. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
