

Mitophagy is required for acute cardioprotection by simvastatin
- PMID: 23901824
- PMCID: PMC4208607
- DOI: 10.1089/ars.2013.5416
Mitophagy is required for acute cardioprotection by simvastatin
Abstract
Aims: We have shown that autophagy and mitophagy are required for preconditioning. While statin's cardioprotective effects are well known, the role of autophagy/mitophagy in statin-mediated cardioprotection is not. In this study, we used HL-1 cardiomyocytes and mice subjected to ischemia/reperfusion to elucidate the mechanism of statin-mediated cardioprotection.
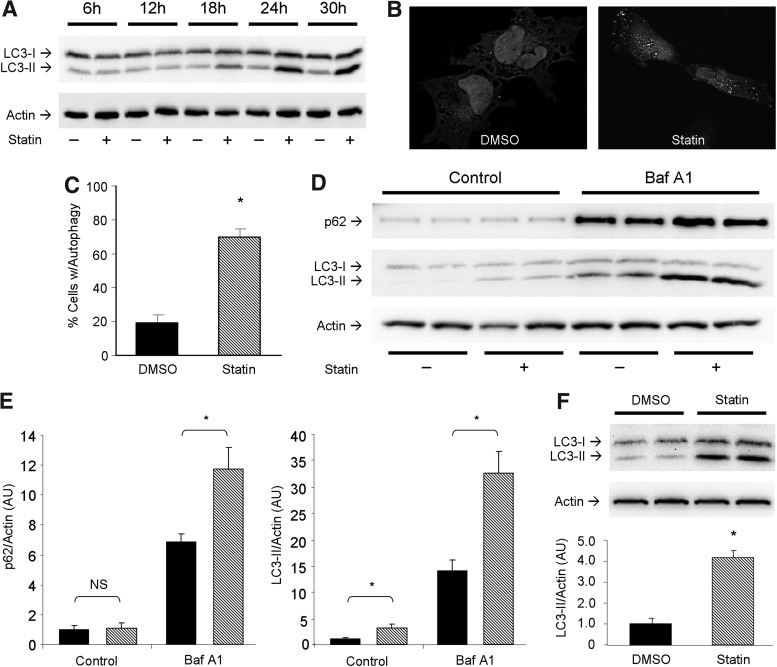
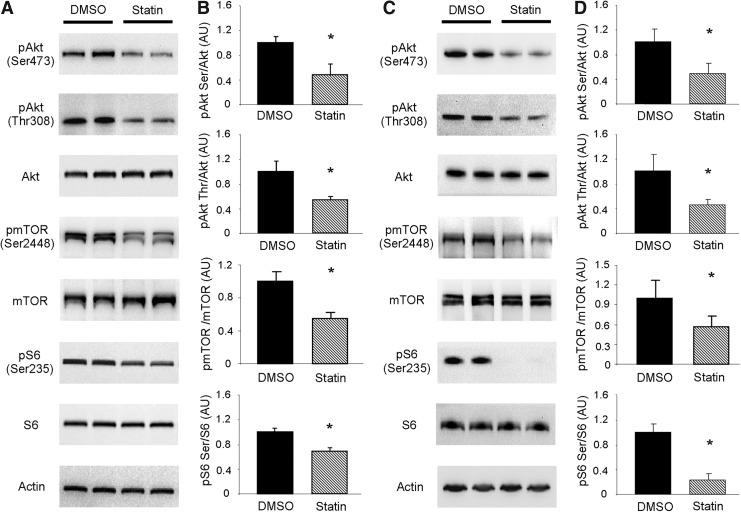
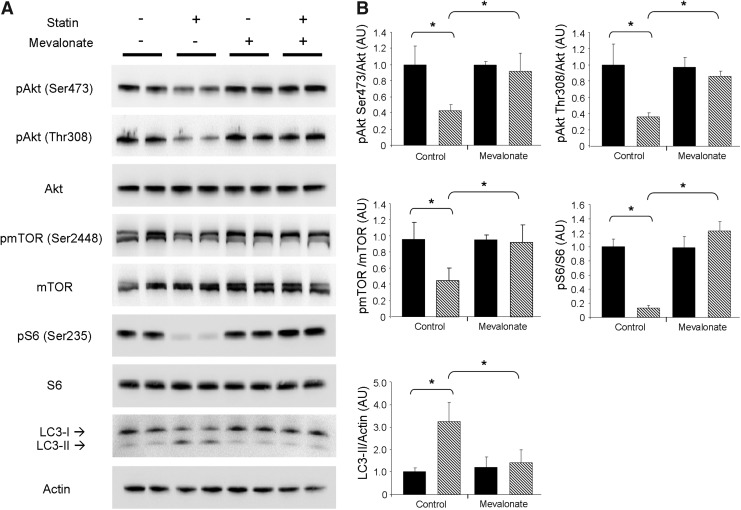
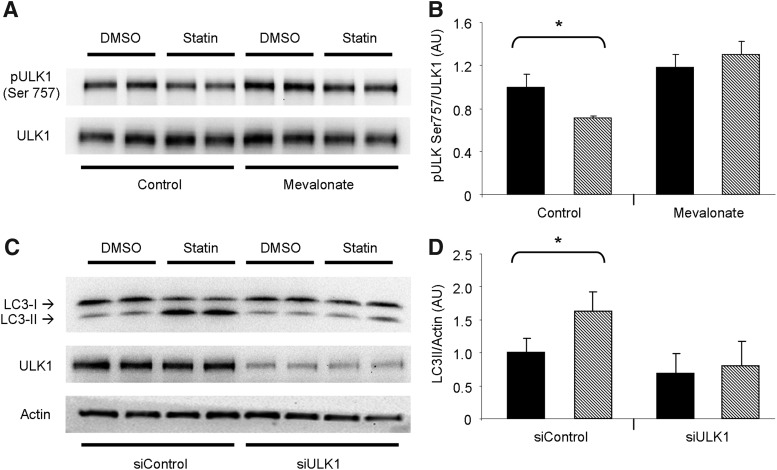
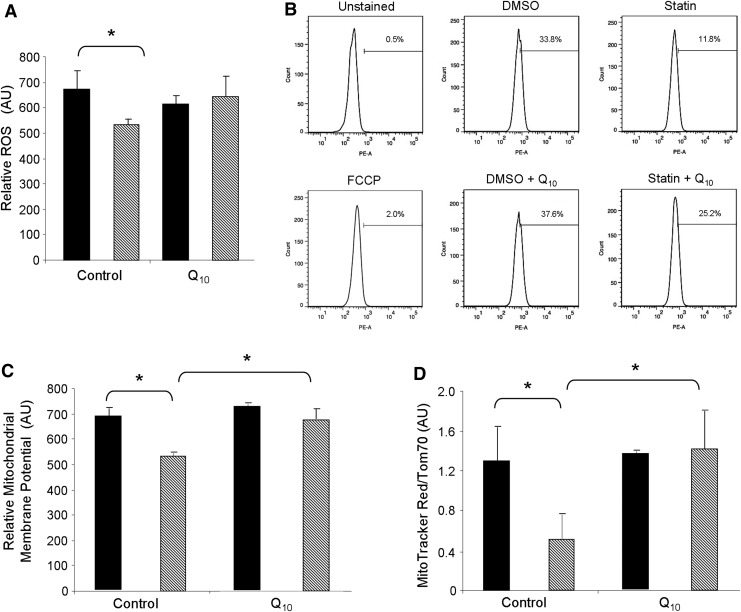
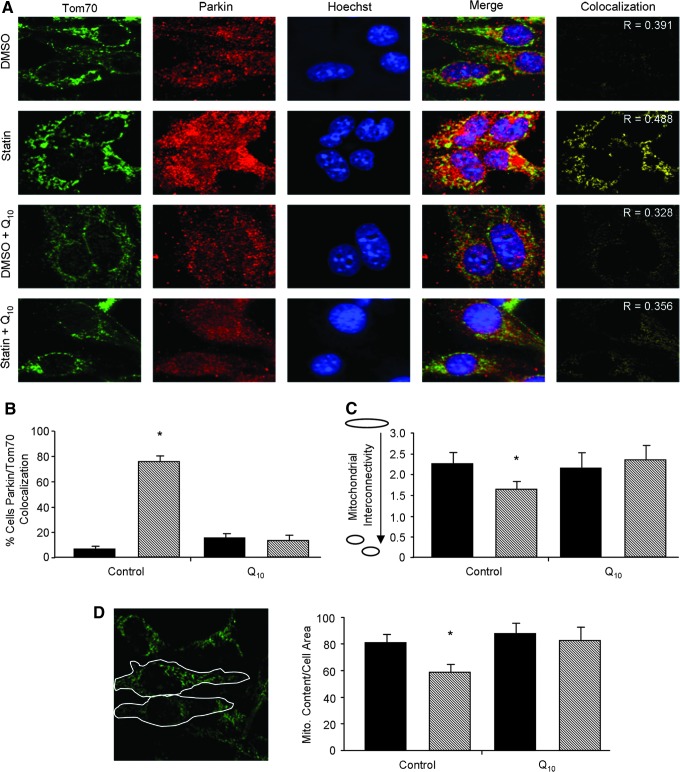
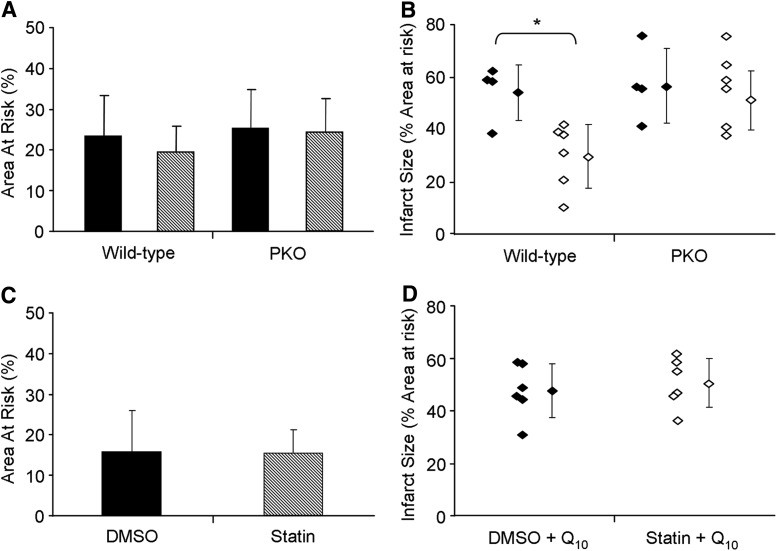
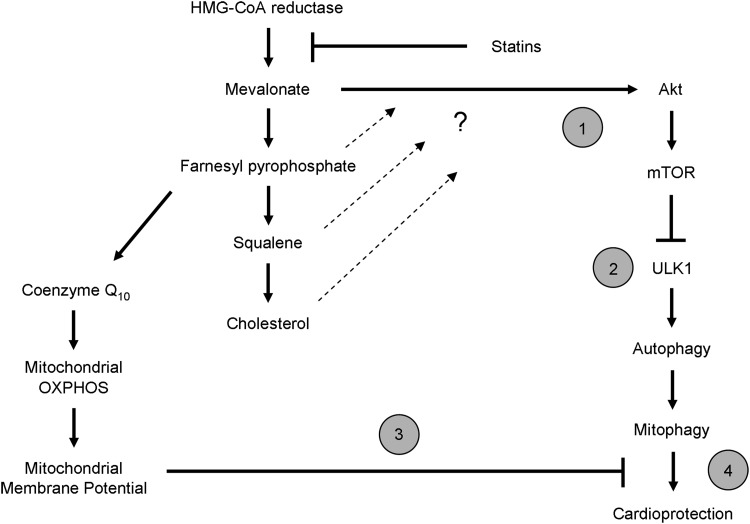
Results: HL-1 cardiomyocytes exposed to simvastatin for 24 h exhibited diminished protein kinase B (Akt)/mammalian target of rapamycin (mTOR) signaling, increased activation of unc-51-like kinase 1, and upregulation of autophagy and mitophagy. Similar findings were obtained in hearts of mice given simvastatin. Mevalonate abolished simvastatin's effects on Akt/mTOR signaling and autophagy induction in HL-1 cells, indicating that the effects are mediated through inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. Simvastatin-treated HL-1 cells exhibited mitochondrial translocation of Parkin and p62/SQSTM1, fission, and mitophagy. Because Parkin is required for mitophagy and is expressed in heart, we investigated the effect of simvastatin on infarct size in Parkin knockout mice. Simvastatin reduced infarct size in wild-type mice but showed no benefit in Parkin knockout mice. Inhibition of HMG-CoA reductase limits mevalonate availability for both cholesterol and coenzyme Q10 (CoQ) biosynthesis. CoQ supplementation had no effect on statin-induced Akt/mTOR dephosphorylation or macroautophagy in HL-1 cells, but it potently blocked mitophagy. Importantly, CoQ supplementation abolished statin-mediated cardioprotection in vivo.
Innovation and conclusion: Acute simvastatin treatment suppresses mTOR signaling and triggers Parkin-dependent mitophagy, the latter which is required for cardioprotection. Coadministration of CoQ with simvastatin impairs mitophagy and cardioprotection. These results raise the concern that CoQ may interfere with anti-ischemic benefits of statins mediated through stimulation of mitophagy.
Figures
References
-
- Araki M, Maeda M, and Motojima K.Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur J Pharmacol 674: 95–103, 2012 - PubMed
-
- Avis HJ, et al. Rosuvastatin lowers coenzyme Q10 levels, but not mitochondrial adenosine triphosphate synthesis, in children with familial hypercholesterolemia. J Pediatr 158: 458–462, 2011 - PubMed
-
- Bentinger M, Dallner G, Chojnacki T, and Swiezewska E.Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radic Biol Med 34: 563–575, 2003 - PubMed
-
- Cafforio P, Dammacco F, Gernone A, and Silvestris F.Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 26: 883–891, 2005 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
