

Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill
- PMID: 23902988
- PMCID: PMC3834857
- DOI: 10.1038/ismej.2013.129
Transcriptional response of bathypelagic marine bacterioplankton to the Deepwater Horizon oil spill
Abstract
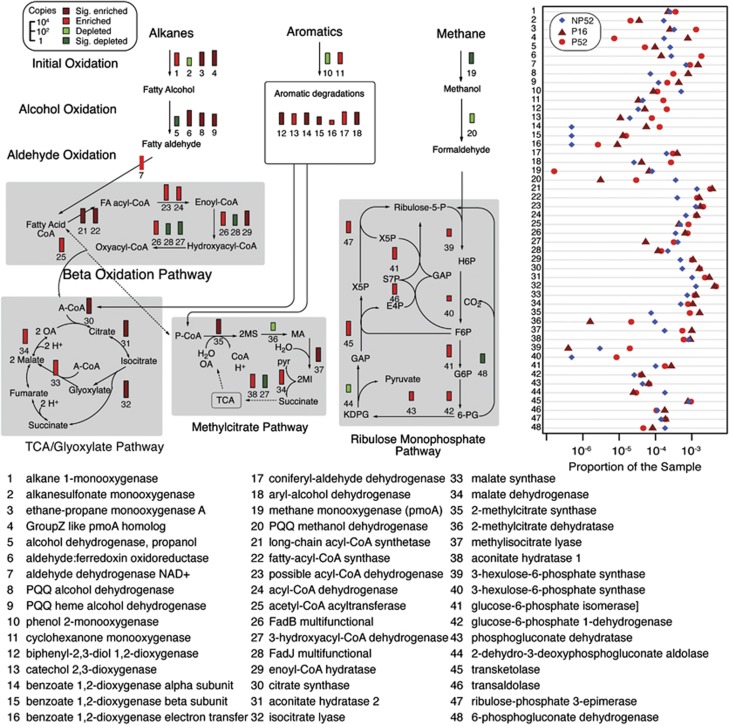
The Deepwater Horizon blowout released a massive amount of oil and gas into the deep ocean between April and July 2010, stimulating microbial blooms of petroleum-degrading bacteria. To understand the metabolic response of marine microorganisms, we sequenced ≈ 66 million community transcripts that revealed the identity of metabolically active microbes and their roles in petroleum consumption. Reads were assigned to reference genes from ≈ 2700 bacterial and archaeal taxa, but most assignments (39%) were to just six genomes representing predominantly methane- and petroleum-degrading Gammaproteobacteria. Specific pathways for the degradation of alkanes, aromatic compounds and methane emerged from the metatranscriptomes, with some transcripts assigned to methane monooxygenases representing highly divergent homologs that may degrade either methane or short alkanes. The microbial community in the plume was less taxonomically and functionally diverse than the unexposed community below the plume; this was due primarily to decreased species evenness resulting from Gammaproteobacteria blooms. Surprisingly, a number of taxa (related to SAR11, Nitrosopumilus and Bacteroides, among others) contributed equal numbers of transcripts per liter in both the unexposed and plume samples, suggesting that some groups were unaffected by the petroleum inputs and blooms of degrader taxa, and may be important for re-establishing the pre-spill microbial community structure.
Figures




References
-
- Anthony C. The biochemistry of methylotrophs. Academic Press: London, UK; 1982.
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300.
-
- Camilli R, Reddy CM, Yoerger DR, Van Mooy BAS, Jakuba MV, Kinsey JC, et al. Tracking hydrocarbon plume transport and biodegradation at Deepwater Horizon. Science. 2010;330:201–204. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
